Abstract
Background: Transcatheter aortic valve implantation (TAVI) and complex percutaneous coronary interventions (PCI) may require cardiac pacing during device delivery, generally requiring the insertion of a temporary pacing lead via an additional venous access site. The purpose-built Electroducer Sleeve device provides direct wire pacing without the need for a temporary venous pacemaker.
Aims: This study assessed the safety of temporary cardiac pacing using the novel sleeve device during PCI.
Methods: This was a multicentre, non-randomised, prospective, first-in-human, single-arm, pilot study. The primary endpoint was analysis of a safety outcome, defined as the occurrence of haematomas or bleeding complications at the device vascular access site. Secondary endpoints included analyses of effectiveness and qualitative outcomes.
Results: Sixty patients (mean age: 77.9±9.6 years) from 4 centres in France were included: 39 (65%) underwent TAVI, and 21 (35%) underwent PCI. Procedures were performed using the sleeve with access through the radial (32 patients; 53.3%) or femoral arteries (26; 43.3%), or the femoral vein (2; 3.3%). Primary endpoint analysis revealed that 2 patients (3.3%) developed EArly Discharge After Transradial Stenting of CoronarY Arteries Study (EASY) grade I/Bleeding Academic Research Consortium (BARC) type I haematomas at the device access site. As a measure of effectiveness, a haemodynamic effect was observed after each spike delivery in 54 patients (90%). Analyses of other secondary endpoints showed that 2 patients (6.3%) presented asymptomatic radial artery occlusion. No allergies were reported.
Conclusions: This first-in-human trial using the Electroducer Sleeve indicated that this novel, purpose-built, temporary pacing device was safe and effective. Larger prospective studies are required to confirm these findings.
Introduction
During percutaneous cardiovascular interventions, such as transcatheter aortic valve implantation (TAVI) and complex percutaneous coronary interventions (PCI), temporary cardiac stimulation may be required to stabilise the heart and allow for optimal positioning of the stent or valve12. Temporary stimulation may also be needed in case of conduction disturbances, for instance for the management of bradycardia or atrioventricular block during TAVI or rotational atherectomy. This stimulation is typically provided by a temporary venous pacemaker (TVP) positioned in the right ventricle. However, the use of a TVP requires an additional venous access point, usually via the femoral vein, and the insertion of a separate stimulation catheter. This procedure may be associated with complications including, in the worst case, cardiac tamponade due to right ventricular perforation13456.
As an alternative, the use of a direct wire pacing (DWP) technique during TAVI has been assessed in both observational and randomised studies. Although DWP during TAVI has generally been found to be safe and effective when compared to conventional right ventricular pacing4789, technical difficulties, as well as potential risks, have prevented this technique from being widely adopted by the medical community. Indeed, in one of the studies, DWP was performed using crocodile clips, with the cathode attached to the distal external end of the guidewire and the anode attached to the incised skin at the insertion site of the arterial sheath in the anaesthetised groin9. This homemade technique, requiring the insertion of subcutaneous needles, may be associated with an increase in the stimulation threshold (mA) and thus stimulation failures, as well as with an increased risk of the patients experiencing electrical sensations, pain, and bleeding.
A purpose-built direct pacing wire has recently been developed to improve the safety and effectiveness of this technique10. The aim of the current study was to assess the safety of a new direct wire unipolar pacing device, the Electroducer Sleeve (Electroducer), for the delivery of temporary cardiac pacing during PCI without the need for a TVP.
Methods
Study design and patient selection
This multicentre, non-randomised, prospective, first-in-human, single-arm, pilot study (ClinicalTrials.gov: NCT04372654) was conducted using data collected from patients undergoing PCI using the Electroducer Sleeve device at 4 sites in France (Institut Cardiovasculaire de Grenoble, Grenoble; Groupe Cardiovasculaire Interventionnel, Clinique Pasteur, Toulouse; Médipôle Lyon-Villeurbanne, Villeurbanne; and the Institut Cardiovasculaire Paris Sud, Hôpital Privé Jacques Cartier, Massy). Data were prospectively collected at hospitalisation prior to the intervention (baseline), during the intervention or at hospital discharge, and at a follow-up visit 1 month after the intervention (M1).
Investigators at the 4 centres were asked to sequentially enrol a total of 60 patients with an indication for PCI or TAVI requiring temporary cardiac stimulation. Only patients who were aged over 18 years and who were capable of understanding the study and providing informed consent were eligible. Exclusion criteria were 1) patients requiring a definitive pacemaker (PM); 2) pregnant or breastfeeding women; 3) patients under judicial protection, tutorship, or curatorship; 4) patients with a negative Allen test or absent radial pulse for radial route access; and 5) patients participating in another interventional clinical trial.
This study was conducted in compliance with the latest version of the Declaration of Helsinki. Data collection and safety monitoring were carried out by a contract research organisation (Cardiovascular European Research Center [CERC]). An evaluation committee was responsible for the review of the scientific and ethical aspects of the study. The committee was established to ensure the safety of the participants and the validity and integrity of the data collected. The members of the committee had no conflicts of interest regarding the device under evaluation and were independent of the sponsors.
Study outcomes and assessments
The primary endpoint was a safety outcome defined as the occurrence of bleeding complications at the access site where the Electroducer Sleeve was inserted, including haematoma formation (EASY classification) and access-site bleeding (BARC classification)1112. Secondary outcomes were defined as 1) the occurrence of radial artery occlusion, at discharge or at M1, diagnosed by Doppler ultrasound of the radial artery and by a reverse Allen test; 2) the occurrence of allergic or cutaneous adverse reactions, at discharge or at M1, identified through clinical examination of the puncture site; 3) the effectiveness of the Electroducer Sleeve device for temporary cardiac stimulation, evaluated from surface electrocardiograms and through assessment of ventricular capture by measuring the haemodynamic effect induced by each spike delivered via the guidewire; 4) the occurrence of femoral artery palpation anomalies at discharge or at M1 in patients for whom arterial access was achieved via the femoral route; 5) the performance of the device, assessed by measuring the stimulation threshold (mA) with an external pacemaker; 6) the impact of using the device on TAVI procedure duration; and 7) pre- and post-procedure assessments of patient pain using the numerical rating scale (NRS), with scores ranging from 0 (no pain) to 10 (worst pain imaginable).
The Electroducer Sleeve
The Electroducer Sleeve is a sterile non-implantable device composed of conductive material that allows the integration of a temporary pacemaker function into the guidewire used during PCI, thus providing a simplified technique for DWP. The guidewire behaves like an intracardiac cathode. The device is composed of a “sleeve” made of a conductive polymer; it has been adapted for use with 6 Fr introducers (length: ≥65 mm) and is connected to an external pacemaker (anode) via a cable. A second cable (cathode) is then connected to this external pacemaker via a crocodile clip attached to the guidewire used during the procedure (Figure 1). The fact that the anode has a large surface and is in contact with the vascular endothelium and the blood system generates a better pacing threshold compared to cutaneous or subcutaneous anodes13.
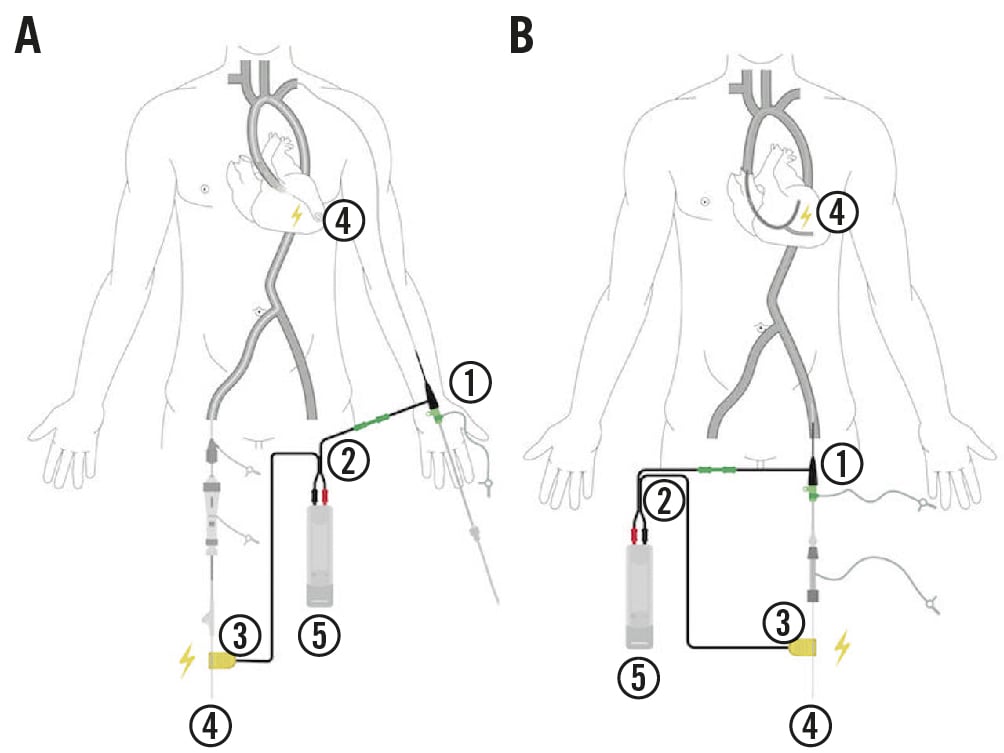
Figure 1. Device set-up according to procedure type. A) Example of a transcatheter aortic valve implantation procedure using the Electroducer Sleeve via the left radial route. B) Example of a femoral percutaneous coronary intervention using the Electroducer Sleeve. 1) Electroducer Sleeve inserted over a 6 Fr introducer; 2) sleeve-clamp-pacemaker connection cable; 3) clamp; 4) ventricular or coronary guidewire; and 5) constant-intensity external pacemaker.
Procedures
All procedures were carried out according to the usual standard practice of each centre. Similarly, the choice of access route (radial or femoral) was left to the discretion of the investigator, as in standard practice, and was based on the profile of the patient (i.e., clinical and anatomical characteristics). All investigators were provided with the information for use (IFU) document for the Electroducer Sleeve, and were instructed to use the DWP device with a constant-current external PM, with adjustable sensitivity and delivering at least 20 mA, and the following guidewires: SION blue (Asahi Intecc), the BMW Universal II (Abbott), the Runthrough NS Floppy (Terumo) and the RotaWire Floppy (Boston Scientific) for coronary interventions, and the SAFARI2 (Boston Scientific), the Amplatz Super Stiff (Boston Scientific), and the Confida Brecker (Medtronic) for TAVI interventions. For all interventions, arterial or venous access was achieved using a 6 Fr introducer (Terumo; length ≥65 mm).
Statistical analysis
Data were analysed using descriptive statistics. Categorical variables were reported as numbers (n) and percentages (%), and continuous variables as the mean±standard deviation (SD) or the median and interquartile range (IQR; Q1 to Q3), as appropriate. As this was an exploratory study, no formal hypothesis was tested.
Results
A total of 60 patients were enrolled in the study from July 2020 to January 2021. Baseline patient and procedural characteristics are reported in Table 1 and Table 2, respectively. The Electroducer device was used in 39 TAVI and 21 PCI procedures. All procedures were successful. No major adverse cardiac and cerebrovascular adverse events (MACCE) were reported during the index hospitalisation or at the M1 follow-up visit.
Regarding the primary endpoint, a total of 2 EASY grade I haematomas, both considered BARC type I, were reported at the puncture site (Table 2). Only one of these haematomas was judged by the investigator as possibly related to the Electroducer Sleeve; this was subsequently confirmed by the contract research organisation and sponsor. The second haematoma was considered to be unrelated to the device. No severe haematomas or bleeding occurred at the device puncture site.
Concerning the secondary outcomes, a total of 2 patients (6.3%) presented radial artery occlusion at the M1 follow-up visit (Table 2). The Electroducer Sleeve was inserted via the femoral artery in 28 patients. Palpation of the femoral artery was carried out at discharge for all 28 patients and at M1 follow-up for 26 of these patients. No femoral artery palpation anomalies were reported during the study. Similarly, no allergic or cutaneous adverse reactions were reported during the procedure or at follow-up.
Overall, regarding the effectiveness endpoint, a haemodynamic effect following each spike delivery was observed in 54 cases (90%). However, stimulation was successful in 56 patients (93.3%). Five of the cases in which no haemodynamic effect for each spike was observed were identified among the first 35 included patients. The patient and procedural characteristics of the cases in which the haemodynamic effect after each spike was not observed are detailed in Supplementary Table 1.
Among the 60 patients, 12 patients underwent a procedure performed with a PM delivering a constant voltage, rather than with a PM delivering a constant current as recommended in the IFU. Four of the cases in which the haemodynamic effect was not observed after each spike occurred in patients who had undergone an intervention using a non-recommended PM (Supplementary Table 1).
For the subpopulation of patients who had undergone an intervention using a recommended constant-current PM, a haemodynamic effect was observed after each spike delivery in 46 cases (95.8%). Stimulation was successful in all of these patients (n=48; 100%), including in the 2 patients for whom a haemodynamic effect after each spike was not observed (Supplementary Table 1).
The mean TAVI procedure duration was 50.8±21.8 minutes. The post-procedure mean NRS score for pain was 0.13±0.47 out of 10, with a score of 0 in 55 of the cases (91.7%) (Table 2). The mean difference in patient pain levels, post- and preprocedure, was 0.08±0.62.
Table 1. Baseline characteristics of the study population.
| Variable | Whole study population N=60 n (%) or mean±SD |
|---|---|
| Age, years | 77.9±9.6 |
| Gender, male | 39 (65.0) |
| BMI, kg/m2 | 27.1±4.2 |
| HTA | 51 (85.0) |
| Diabetes | 17 (28.3) |
| Type I | 2 (11.8) |
| Type II | 15 (88.2) |
| Dyslipidaemia | 33 (55.0) |
| Family history of <60 CAD | 6 (10.0) |
| Current smoker | 6 (10.0) |
| Previous MI | 7 (11.7) |
| Previous TIA/stroke | 2 (3.3) |
| Previous PCI | 29 (48.3) |
| Previous cardiac surgery | 5 (8.3) |
| Pacemaker | 9 (15.0) |
| Previous TAVI | 1 (1.7) |
| Peripheral vascular disease | 5 (8.3) |
| BMI: body mass index; CAD: coronary artery disease; HTA: hypertension; MI: myocardial infarction; PCI: percutaneous coronary intervention; SD: standard deviation; TAVI: transcatheter aortic valve implantation; TIA: transient ischaemic attack | |
Table 2. Procedural characteristics.
| PROCEDURAL CHARACTERISTICS | n (%) | |
|---|---|---|
| Procedure type | n=60 | |
| TAVI | 39 (65.0) | |
| PCI | 21 (35) | |
| Ostial coronary lesions | 11 (18.3) | |
| Rotational atherectomy | 8 (13.3) | |
| Acute coronary syndrome | 1 (1.7) | |
| Complex PCI | 1 (1.7) | |
| Access type | n=60 | |
| Femoral artery | 26 (43.3) | |
| Right | 14 (23.3) | |
| Left | 12 (20.0) | |
| Radial artery | 32 (53.3) | |
| Right | 27 (45.0) | |
| Left | 5 (8.3) | |
| Femoral vein | 2 (3.3) | |
| Type of guidewire used for direct wire pacing | n=60 | |
| Boston Scientific, Amplatz Super Stiff | 9 (15.0) | |
| Boston Scientific, SAFARI2 | 26 (43.3) | |
| Medtronic, Confida Brecker | 4 (6.7) | |
| Asahi, SION blue | 9 (15.0) | |
| Boston Scientific, RotaWire Floppy | 8 (13.4) | |
| Terumo, Runthrough Floppy | 1 (1.7) | |
| Abbott, BMW Universal II | 3 (5.0) | |
| Study outcomes | ||
| Bleeding complications at the sleeve puncture sitea | n=60 | |
| Haematoma | EASY I/BARC I | 2 (3.3) |
| EASY >I/BARC >I | 0 | |
| Major bleeding | BARC >I | 0 |
| Pain assessment | n=60 | |
| NRS=0 | 55 (91.7) | |
| NRS 1-3 | 5 (8.3) | |
| NRS >3 | 0 | |
| Allergic or adverse tissue reactions | 0 | |
| Radial artery occlusion at 1 month, n=32 | 2 (6.3) | |
| aOne patient had BARC type II (EASY grade II) femoral bleeding, and one patient had BARC type IIIa (EASY grade II) femoral bleeding, neither of which occurred at the sleeve puncture site. BARC: Bleeding Academic Research Consortium classification; EASY: EArly Discharge After Transradial Stenting of CoronarY Arteries Study classification; NRS: numerical rating scale; PCI: percutaneous coronary intervention; TAVI: transcatheter aortic valve implantation | ||
Discussion
This multicentre, prospective, single-arm, first-in-human, pilot study using the Electroducer Sleeve, a dedicated DWP device, indicated that the device was safe and effective for temporary cardiac pacing during percutaneous cardiovascular interventions. Use of the device was not associated with the occurrence of severe bleeding events or haematomas at the vascular access site, and radial artery occlusion and femoral artery palpation anomalies were uncommon. The device was also well tolerated with no allergies or cutaneous adverse events being reported. Finally, patient-reported pain assessment scores were very low.
This pilot study was conducted to provide a thorough assessment of the safety and performance of the Electroducer Sleeve. Thus, the primary endpoint for this first-in-human study was based on the analysis of a safety outcome to assess the occurrence of device-related haematomas and bleeding. Our results indicated that the rate of haematomas induced by the Electroducer Sleeve was inferior to that reported historically for puncture-site complications during percutaneous cardiovascular interventions. In our study, minor haematomas (classified as EASY grade I/BARC type I) occurred in only 2 patients (3.3%) at the sleeve puncture site (1 femoral haematoma and 1 radial local haematoma). Previous studies have suggested that haematoma incidence varies from 9.4% for interventions involving radial access up to 17.4% for those involving femoral access1114. In addition, radial occlusion, evaluated by Doppler ultrasound, was observed in 2 patients (6.3%) in our study. Historically, the rates of radial occlusion described in literature have been reported to range from 2% to 15% depending on the radial compression technique used (i.e., manual, use of a radial compression device, or the patent haemostasis technique)1516. Indeed, in both cases of radial occlusion occurring in our study, the compression times were long, exceeding 6 hours. A study in a contemporary setting with a larger cohort is therefore needed to evaluate the specific issue of the occurrence of radial artery occlusion during DWP.
Overall, each spike delivery was followed by a haemodynamic effect in 54 of the patients (90%). However, the analysis of this outcome was judged to be inaccurate for 12 patients from one of the centres; in these patients, the procedure had been performed using a type of pacemaker that was not recommended by the IFU document provided for the sleeve device. Indeed, according to Ohm’s law (V=R*I), unipolar pacing intensity depends on the patient’s body impedance, unlike in bipolar pacing where impedance is fixed. In clinical practice, a patient’s body resistance (measured in Ohms, Ω) is unknown and can vary considerably (by 1- to 5-fold) depending on various factors such as body hydration status, body mass index, cardiac failure, hydrops, and the presence of myocardial scars. Thus, voltage pacing settings on the device do not allow the current intensity which is actually delivered to the myocardium to be predicted. The IFU for the sleeve device recommends regulating current intensity in milliamps (mA) rather than in volts (V), and therefore the use of a constant-current external PM with an output of at least 20 mA and adjustable sensitivity (which includes most modern and widely available PM) during interventions using the Electroducer Sleeve ensures the compatibility of the external PM with unipolar stimulation. Among the patients who had undergone procedures using a recommended constant-current PM (48 patients), loss of capture occurred in only 2 patients, compared to in 6 patients in the whole study population. Loss of capture is a frequent event that can be caused by multiple factors, including stimulation in an infarcted area, incorrect clamp or guidewire positioning, low levels of patient hydration, and spike delivery during the refractory period17. This event is frequently reported in the United States Food and Drug Administration Manufacturer and User Facility Device Experience (MAUDE) database and may not be specifically related to the pacing device being used. Moreover, a mean rate of transvenous temporary pacing failure of 9.5% was reported in a scoping literature review5. The loss of capture rate observed using the study device was similar to that observed in standard practice using the DWP technique. In the randomised EASY TAVI study, effective stimulation rates were similar between the group stimulated using a standard catheter and the DWP group: 124 patients (84.9%) versus 128 (87.1%), respectively (p=0.60)9. Based on these data, the effectiveness of the Electroducer Sleeve can be considered to be similar to that of other pacing devices, including those using a standard pacing lead and those using the DWP technique.
The average TAVI procedure duration observed in our study (50.8±21.8 min) was similar to that observed in the EASY TAVI study (55.6±26.9 min using a standard pacing lead and 48.4±16.9 min using the DWP technique), indicating that the Electroducer Sleeve does not increase procedure times. Finally, our study showed that the Electroducer Sleeve was well tolerated, as indicated by the low NRS pain scores and the absence of any cutaneous adverse reactions.
Limitations
As this was a pilot first-in-human study of a new medical device, patient safety was a major priority and, thus, only a limited number of patients were included. Therefore, our study provides only exploratory data and lacks the power needed to determine the statistical significance of the main safety analysis or of the effectiveness endpoint. In addition, the trial was not randomised, and the lack of a control population prevented a definitive evaluation of the efficacy of the Electroducer Sleeve device. However, assessment of the effectiveness of the device was carried out using strict criteria, involving measurement of the haemodynamic effect induced by each spike rather than assessment of the global haemodynamic effect used in previous studies3718. In addition, this multicentre and prospective study did not identify any safety concerns related to bleeding at the puncture site or any other complications. Further studies are now needed to confirm our preliminary findings.
Conclusions
This first-in-human study of the use of a new purpose-built DWP device indicated that the device was safe, effective and well tolerated by the patients. Larger prospective studies are required to confirm these findings and for detailed evaluations of device efficacy.
Impact on daily practice
The Electroducer Sleeve is a novel, purpose-built device that appears to provide safe and effective unipolar pacing during percutaneous cardiovascular interventions, without the need for a temporary venous pacemaker.
Acknowledgements
Medical writing assistance and language editing were provided by Drs Emma Pilling and Marielle Romet (Synpharm-Santé Active Edition).
This paper was guest edited by Franz-Josef Neumann, MD; Department of Cardiology and Angiology II, University Heart Center Freiburg - Bad Krozingen, Bad Krozingen, Germany.
Funding
The study was funded by Electroducer and the Fondation de l’Avenir.
Conflict of interest statement
The authors have no conflicts of interest to declare. The Guest Editor reports lecture fees paid to his institution from Amgen, Bayer Healthcare, Biotronik, Boehringer Ingelheim, Boston Scientific, Daiichi Sankyo, Edwards Lifesciences, Ferrer, Pfizer, and Novartis; consultancy fees paid to his institution from Boehringer Ingelheim; and grant support from Bayer Healthcare, Boston Scientific, Biotronik, Edwards Lifesciences, GlaxoSmithKline, Medtronic, and Pfizer.
Supplementary data
To read the full content of this article, please download the PDF.
