Introduction
The tricuspid valve is often referred to as the “forgotten valve”. Tricuspid valve disease, in particular moderate-to-severe tricuspid regurgitation (TR), is present in over 1.5 million US individuals and frequently co-exists with left-sided valve disease. Despite clinical guidelines recommending concomitant tricuspid valve repair at the time of left-sided valve surgery, this is seldom undertaken when moderate-severe TR is present. Furthermore, isolated tricuspid valve repair late following left-sided valve surgery entails significant clinical risk, with high mortality rates. As such, the significant burden of residual tricuspid disease in high surgical risk patients has spawned the evolution of a number of percutaneous treatment options. These include percutaneously delivered annular rings, transcatheter suture bicuspidisation, devices to improve lealfet coaptation, and valved stents or heterotopically placed transcatheter aortic valves within the vena cava.
The FORMA Repair System
DEVICE CONCEPT AND DESIGN
The FORMA Repair System (Edwards Lifesciences, Irvine, CA, USA) represents a novel, fully percutaneous treatment option for patients with moderate-severe TR. The chief design element involves placing a device to reduce the regurgitant orifice area (and thus the amount of TR), thereby providing a surface for valve leaflet coaptation. The device consists of a spacer (Figure 1A) and a rail (Figure 1B), which is anchored within the right ventricular apex (Figure 1C). The spacer consists of a foam-filled polymer balloon that passively expands via holes within the spacer shaft. There are currently two available coaptation diameter sizes (12 mm and 15 mm). Overall spacer length is 42 mm with radiopaque markers at each end of the spacer. The device is positioned such that the spacer occupies the regurgitant space and is ultimately fixed in the distal right ventricular myocardium. The fixation mechanism consists of a six-pronged nitinol anchor, designed to minimise the risk of perforation.
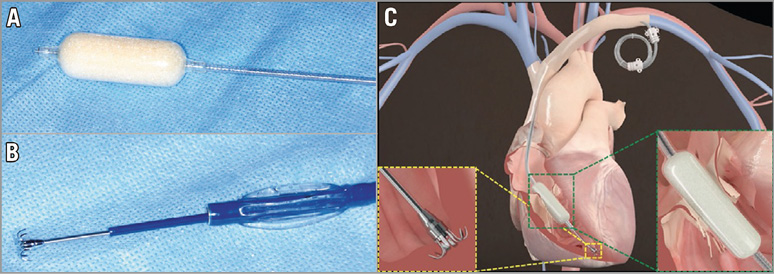
Figure 1. The FORMA Repair System. A) Spacer. B) Steerable delivery catheter and anchoring system. C) Device at the tricuspid valve annulus, anchoring system at the right ventricular apex, and excess device length coiled into a subcutaneous pocket. Reproduced with permission from Campelo-Parada et al, J Am Coll Cardiol. 2015;66:2475-83.
KEY POINTS FOR PATIENT SELECTION
Clinical indications currently include severe TR in the presence of symptomatic right heart failure deemed by the Heart Team to be at very high or prohibitive surgical risk. Patients with prior tricuspid valve surgery have been excluded. Transthoracic and transoesophageal echocardiography, along with computed tomography, are essential for pre-procedural patient selection and planning. Key issues involve assessing not only the severity and mechanism of TR, annular dimensions, and right ventricular geometry, but also the status of subclavian venous access to ensure an adequate size to facilitate the introducer sheath and device.
PROCEDURAL STEPS
Fluoroscopic guidance (Figure 2) coupled with 2D and 3D transoesophageal echocardiography is utilised. Following left axillary venous access, a large bore sheath (20 Fr for the 12 mm-sized device, 24 Fr for the 15 mm-sized device) is secured to accommodate the spacer. Right ventriculography is performed to locate fluoroscopically the tricuspid annular plane and true right ventricular apex (Figure 2A). Ideally, the device should be positioned perpendicular to the valve plane to ensure optimal device coaptation. The anchor site is aimed at the RV wall perpendicular to the centre of the annulus. A steerable delivery catheter is positioned within the right ventricle for rail system delivery (Figure 2B, Figure 2C). The spacer is then tracked over the rail to the tricuspid valve plane and placed in the best position to reduce TR, assessed live with echocardiography. The device is then locked proximally, and the excess rail length is coiled and placed within a subcutaneous pocket. If required, the entire device is fully retrievable during all stages of the procedure until sheath removal.
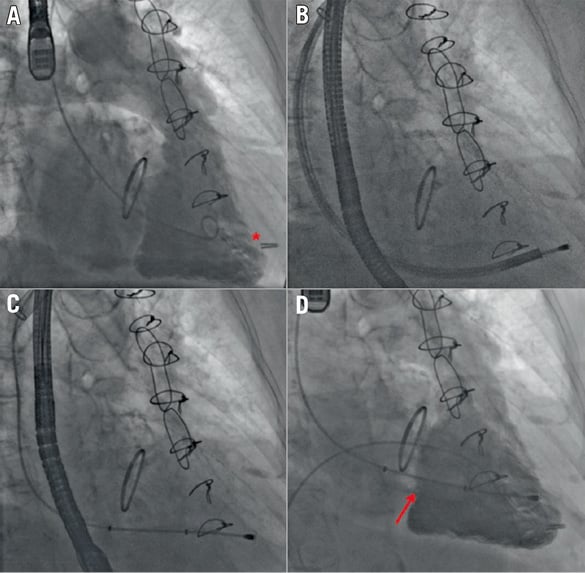
Figure 2. Intraprocedural fluoroscopic/angiographic images. A) Right ventriculography to locate the tricuspid annular plane and identify the ideal anchor location (red asterisk) on fluoroscopy. B) Right ventricular anchoring via a steerable delivery catheter. C) Device positioning in the valve plane. D) Final right ventriculography showing the device in correct position (red arrow) and reduction of tricuspid regurgitation from baseline. Reproduced with permission from Campelo-Parada et al. J Am Coll Cardiol. 2015;66:2475-83.
First-in-man results
BASELINE FEATURES
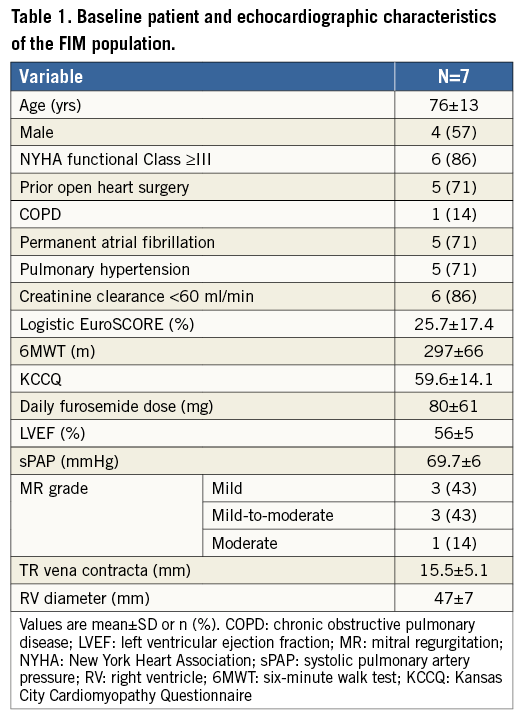
The first-in-man experience involved seven patients (mean age 76±13 yrs) who underwent FORMA device implantation under a compassionate clinical use programme and was recently reported. The main baseline characteristics of this cohort of patients are shown in Table 1. The mean logistic EuroSCORE was 25.7±17.4%, mean left ventricular ejection fraction was 56±15% and New York Heart Association (NYHA) functional class across most patients was III or IV. Pulmonary hypertension was evident in five patients, and five patients had prior cardiac surgery. Renal insufficiency and permanent atrial fibrillation were also observed in most of these patients. The mean baseline furosemide dose was 80±61 mg per day. From an imaging standpoint, mean maximal vena contracta measured 15.5±5.1 mm, with severe leaflet non-coaptation being noted in all of these patients with consequent severe TR.
PROCEDURAL AND IN-HOSPITAL RESULTS
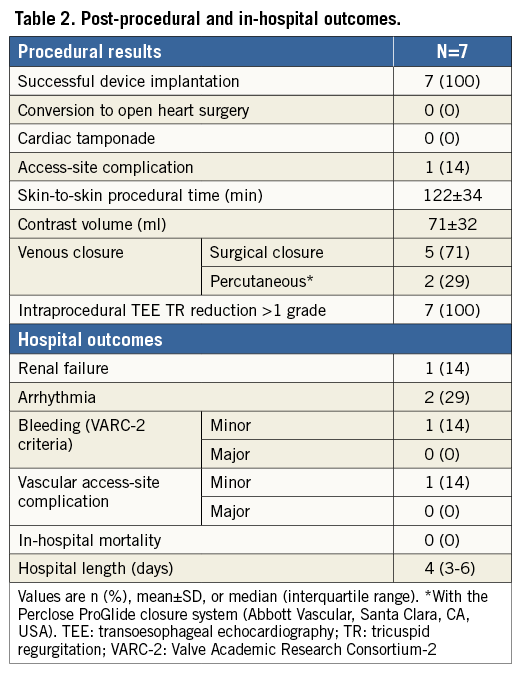
Successful device implantation occurred in all patients without procedural complications (Table 2). The severity of TR assessed intraprocedurally was reduced by at least one grade in all patients, with four patients demonstrating reductions of two TR grades. One patient experienced several episodes of asymptomatic non-sustained (right ventricular outflow tract) ventricular tachycardia within the 24 hours post-procedure, which was subsequently controlled with beta-blockers. Another two patients demonstrated frequent premature ventricular contractions within the first 24 hours post-procedure. One patient experienced minor bleeding related to vascular access. Median length of hospital stay was four (three to six) days. All but one patient received warfarin post-procedure.
30-DAY RESULTS
All patients were alive at 30-day follow-up, without further complications. Improvements in NYHA functional class were observed in all but one patient, along with significant reductions in quantity of peripheral oedema. This also coincided with reductions in diuretic dosages in two patients. From an imaging standpoint, TR was assessed as being moderate in severity in all patients, without evidence of tricuspid stenosis. Despite the relatively short time period from device implantation, quality of life improvements were noted in all of the four patients who undertook the Kansas City Cardiomyopathy Questionnaire (KCCQ) (59.6±14.1 to 86.2±5.4), and exercise capacity also increased in 4/5 patients who performed the six-minute walk test (297±66 m to 326±74 m).
Future perspectives
Preliminary six-month data from an updated first-in-man cohort of 14 patients who underwent FORMA device implantation were recently reported. Successful device implantation was obtained in all but one patient, and all patients were alive at 30 days. The severity of TR was reduced in all but one patient, and at six-month follow-up 12 patients (86%) were in NYHA functional Class I-II.
An early feasibility study is currently ongoing within the USA (NCT02471807). It is planned to enrol 15 patients in the study and the primary outcome is procedural success, defined as successful device implantation with no device or procedural serious adverse events at 30 days. A larger trial will be launched in the second half of 2016 (SPACER trial, NCT02787408). The estimated enrolment consists of 75 patients, and the primary outcome will assess the cardiac mortality of the as treated cohort at 30 days compared to a literature-derived performance goal based on high-risk surgical outcomes for tricuspid repair/replacement.
Summary
A percutaneous technique for reducing severe TR using the FORMA Repair System appears safe, and technically feasible. First-in-man data have demonstrated an acute intraprocedural reduction in TR severity in the vast majority of cases, coupled with evidence of functional improvement in the short to medium term. The ongoing registries will enrol larger patient numbers with longer follow-up periods to understand device performance better. Ultimately, randomised trials will be needed to provide definite data on the efficacy of the FORMA device for the treatment of this high-risk group of patients with severe symptomatic TR.
Conflict of interest statement
J. Rodés-Cabau reports having received research grants from Edwards Lifesciences. R. Puri has no conflicts of interest to declare.
