Abstract
Aims: The EVOLVE FHU trial demonstrated non-inferiority of six-month late loss with two dose formulations of SYNERGY, a novel bioabsorbable polymer everolimus-eluting stent (EES) compared with the durable polymer PROMUS Element (PE) EES. The current analysis describes the six-month IVUS and clinical results through two years from the EVOLVE FHU trial.
Methods and results: EVOLVE recruited 291 patients from 29 centres. At six months, IVUS-assessed in-stent net volume obstruction was 3.40±5.06% for PROMUS Element (PE) vs. 2.68±4.60% for SYNERGY (p=0.34) and 3.09±4.29% for SYNERGY ½ dose (p=0.68 vs. PE). There were no significant differences between groups for any other measured IVUS parameter including resolved, persistent, and late-acquired incomplete stent apposition (ISA). At two years, target lesion failure (TLF) was 6.1% for PE vs. 5.5% for SYNERGY (p=0.87) and 5.2% for SYNERGY ½ dose (p=0.81). There were no significant differences between groups for cardiac death, repeat revascularisation, MI or stent thrombosis through two years.
Conclusions: At six months, everolimus delivered from an ultrathin bioabsorbable abluminal polymer resulted in equivalent net volume obstruction and ISA compared with a permanent polymer EES. There were no significant differences between PE and either SYNERGY stent for any major cardiac endpoint through two years. Clinical trials number: NCT01135225.
Introduction
Over the last ten years, there have been limits to the success of DES, most notably due to the concern over late stent thrombosis and patient compliance with dual antiplatelet therapy1,2. The underlying causes of stent thrombosis (ST) have not been fully elucidated and are likely multifactorial - dependent on patient, lesion, underlying disease and procedural factors. However, it has been shown that early discontinuation of clopidogrel increases the risk of ST and longer thienopyridine compliance can decrease the incidence of late ST3-5.
Recent animal and human studies suggest that the durable polymer may be a potential source of ST due to prolonged or chronic inflammation and hypersensitivity reactions3,6-13. DES can delay vessel healing resulting in impaired stent strut coverage which may be associated with late ST demonstrated in human autopsy studies10. Therefore, DES design has progressed toward reduced polymer load with short-term exposure and minimal drug burden. SYNERGY is a novel thin strut platinum chromium stent with an ultrathin, bioabsorbable, everolimus-eluting, abluminally-applied polymer coating14. Once the drug and polymer are resorbed, all that is left behind is a bare metal scaffold.
The EVOLVE FHU trial compared two dose formulations of SYNERGY to the permanent polymer PROMUS Element everolimus-eluting stent (PE). At six-months, the SYNERGY stent was found to be non-inferior to PE for the primary angiographic endpoint of in-stent late loss. Clinical events were low and similar between groups at six months with no ST in any group. The current analysis describes the six-month intravascular ultrasound (IVUS) and clinical results through two years from the EVOLVE trial.
Methods
The EVOLVE FHU (first human use) trial has been previously described14 and is briefly summarised here.
Device description
The PROMUS Element (PE) stent elutes everolimus from a conformal permanent polymer coating on a platinum chromium alloy scaffold15. The SYNERGY stent uses an ultrathin abluminal, bioabsorbable, everolimus-eluting poly(DL-lactide-co-glycolide) (PLGA) coating designed to resorb shortly after drug elution is complete at three months. The everolimus dose is similar to that of PROMUS Element (1 μg/mm2). The SYNERGY ½ dose stent uses the same coating and scaffold as the SYNERGY stent but one-half the dose of everolimus. The cumulative release profile of everolimus from the SYNERGY stent is similar to PROMUS Element16.
Subject selection, procedure and follow-up
Patients with de novo native coronary lesions ≤28 mm in length, reference vessel diameter ≥2.25 mm but ≤3.5 mm, and percent diameter stenosis >50 were randomised single-blind 1:1:1 at 29 sites in the European Union, New Zealand, and Australia to receive either SYNERGY, SYNERGY ½ dose, or PE. Patients with left main disease, chronic total occlusion, acute MI or recent MI were excluded. We have previously reported that both SYNERGY stents were non-inferior to PE for the primary endpoint of in-stent six-month late loss. Thirty-day target lesion failure (TLF: target vessel-related MI, target vessel-related cardiac death, or target lesion revascularisation [TLR]) was similar between groups14. Lesions were evaluated by IVUS and angiography at six months post-procedure, and clinical follow-up occurred at one and two years. Clinical follow-up will continue annually through five years. Beyond the 12-month time point, follow-up was limited to those patients who received a study stent. Patients who are enrolled but who did not receive a study stent were followed for 12 months. IVUS parameters assessed included percent net volume obstruction, incomplete apposition, stent/lumen/vessel/neointimal areas and volumes.
IVUS
IVUS was required post-procedure and six months post-index-procedure for patients who received a study stent. IVUS was performed according to a standardised protocol using a motorised pullback throughout the stented segment and references and following established guidelines17. IVUS images were digitally collected and sent to the IVUS Core Laboratory for central, blinded analysis (MedStar Health Research Institute, Washington DC, USA) employing standard measures and analysis17. Assessors were blind to study treatment. All IVUS image analysis was performed and reviewed by experienced personnel using commercially approved equipment (Indec Medical Systems, Santa Clara, CA, USA). Post-procedure and follow-up images were aligned by visualising the stent segment. Only images with motorised pullbacks at both time points and in which the stent length on the IVUS images were matched as well were used for volumetric analysis17.
Study endpoints
The primary angiographic endpoint was in-stent late loss as measured by an independent core-laboratory adjudicated QCA at six months14. The primary clinical endpoint was target lesion failure (TLF), a composite of cardiac death or MI related to the target vessel, or ischaemia-driven target lesion revascularisation (TLR) at 30 days14. The IVUS endpoints measured by an independent core laboratory included post-procedure and six-month % net volume obstruction, incomplete apposition and stent/lumen/vessel/neointimal areas as well as volumes. Other clinical endpoints have been described previously14.
Statistical methods
This trial was powered for testing of non-inferiority for the six-month primary angiographic endpoint; subsequent analyses should be considered post hoc and exploratory in nature. To control systematic error or bias, patients will remain blinded to treatment throughout the course of the study. Values are expressed as mean ± standard deviation (SD) for continuous variables and percent (count/sample size) for discrete variables. Statistical significance was set at p<0.05 and was determined by a two-sided Student t-test for continuous variables and chi-square or Fisher exact test for discrete variables. The chi-square test was used by default; the Fisher exact test was used when the total number of samples ≤40 and/or at least one cell count in the 2 by 2 table had an expected value <5. Survival curves were constructed and outcomes at two years are presented for time-to-event variables using Kaplan-Meier estimates. Analyses were performed using SAS version 8.2 or higher (SAS Institute, Cary, NC, USA).
Results
Baseline characteristics of the randomised arms of the EVOLVE FHU study have been previously published14. The number of patients available for follow-up through two years for each randomised group is shown in Figure 1. Follow-up for IVUS was 94.2% (274/291) post-procedure and 88.3% (257/291) at six months. Patients were not included in the IVUS analysis because they could not attend the visit (n=13), unknown reason (n=10), equipment/database error (n=6), out of medical necessity (n=3) or did not receive a study stent (n=2). Follow-up for 12-month and two-year clinical endpoints was 97.9% (285/291) and 97.2% (281/289), respectively (Figure 1).
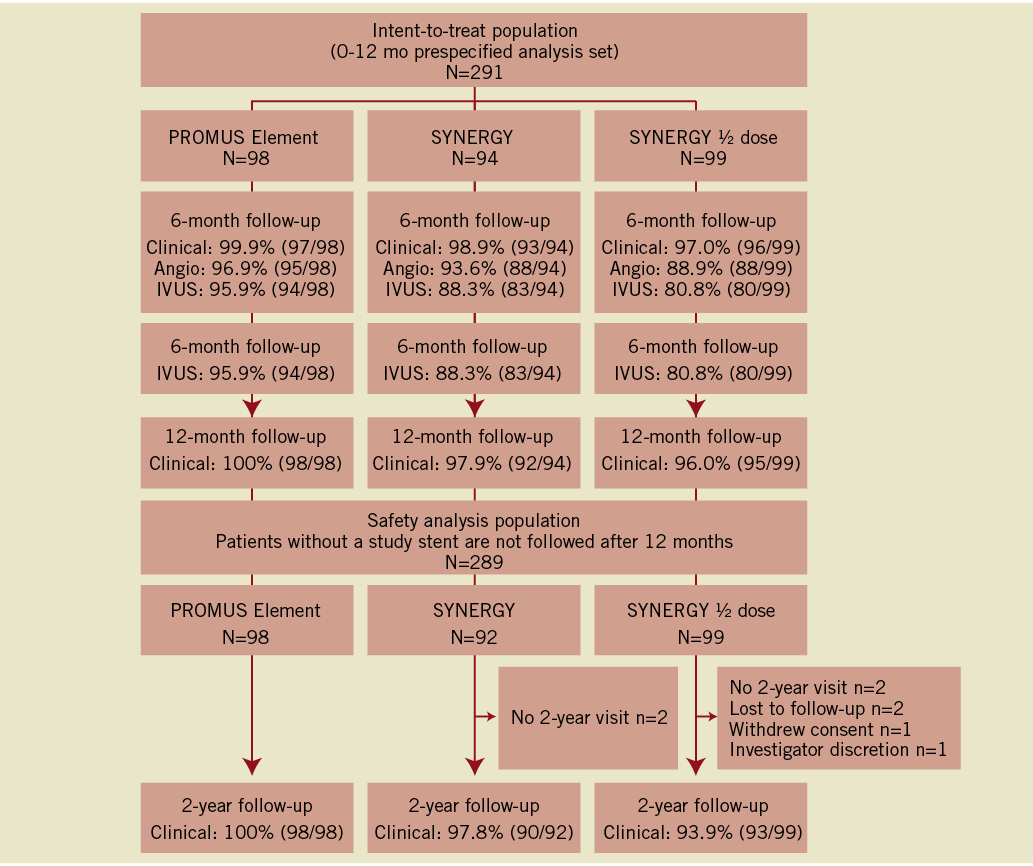
Figure 1. Patient disposition to two years. Patients were analysed in an intent-to-treat manner up to and including the 12 month time point. At the two-year follow-up, the prespecified safety analysis patient population was used, which included only those patients treated with a study stent. Two SYNERGY patients were not included in this safety analysis as they did not receive the study stent.
IVUS results at six months
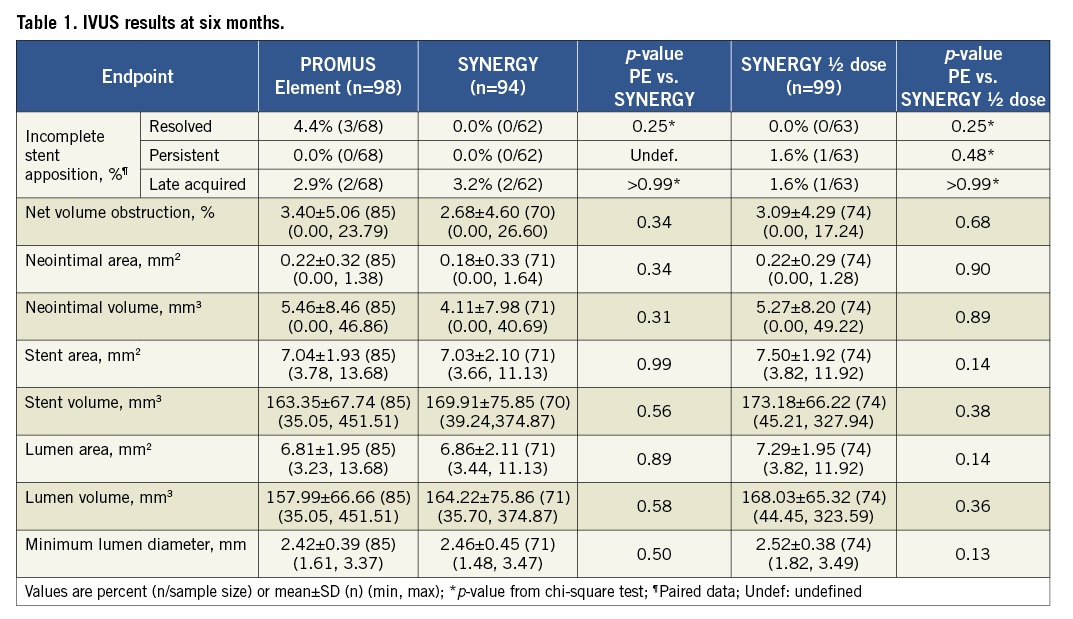
At six months, in-stent net volume obstruction was 3.40±5.06% for PE vs. 2.68±4.60% for SYNERGY (p=0.34) and 3.09±4.29% for SYNERGY ½ dose (p=0.68 vs. PE) (Table 1). Resolved, persistent and late-acquired incomplete stent apposition (ISA) were, respectively, 4.4%, 0%, and 2.9% for PE, 0%, 0%, and 3.2% for SYNERGY, and 0%, 1.6% and 1.6% for SYNERGY ½ dose (Table 1). There were no significant differences between groups for any other measured IVUS parameters (Table 1). No significant differences were found in the change of any IVUS outcome from post-procedure to six months (PE n=68, SYNERGY n=62, SYNERGY ½ dose n=63; Online Figure 1).
Clinical results
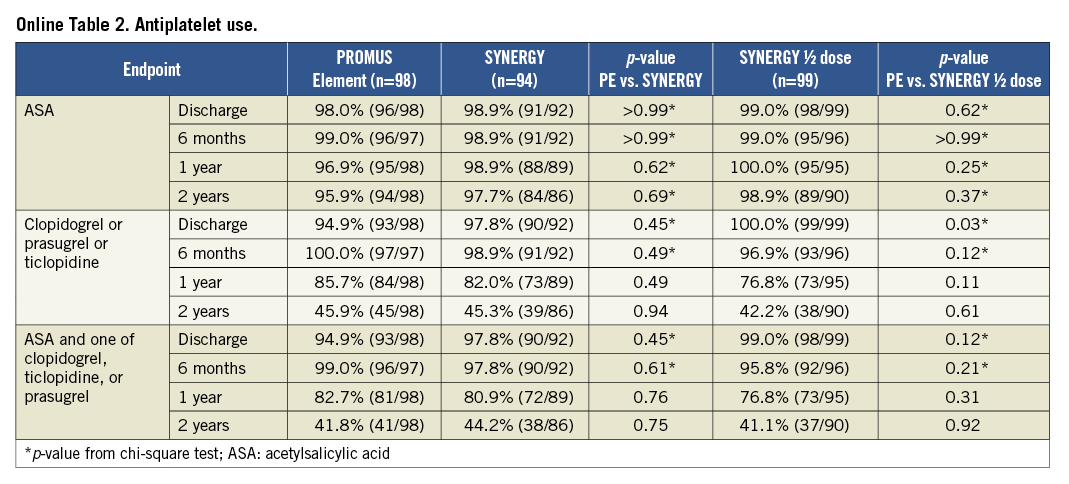
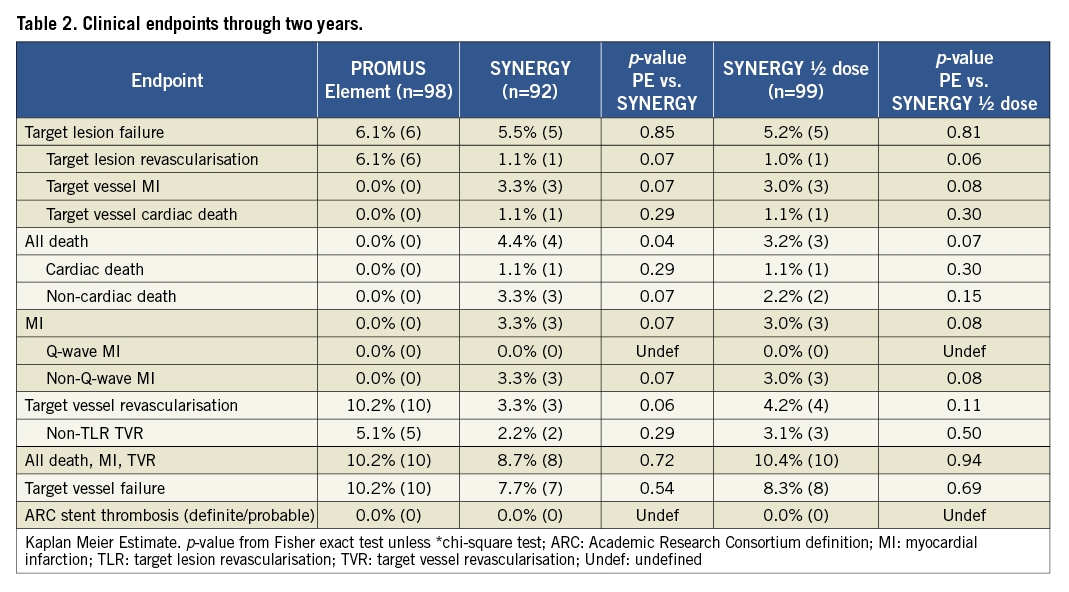
At 12 months, TLF was 5.1% for PE vs. 4.4% for SYNERGY (p>0.99) and 4.2% for SYNERGY ½ dose (p>0.99). There were no significant differences between groups for any clinical endpoint at 12 months, including death, repeat revascularisation and MI. At discharge, significantly more patients in the SYNERGY arms were taking antiplatelet therapy compared with PE.
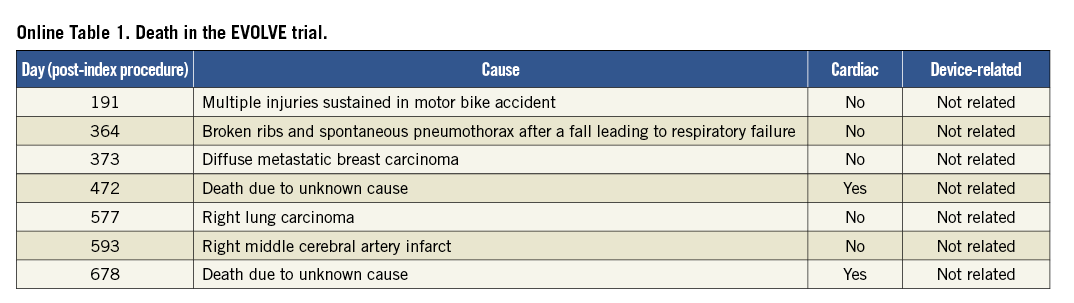
After an additional year of follow-up, TLF was 6.1%, 5.5% and 5.2% in the PE, SYNERGY and SYNERGY ½ dose arms, respectively (p=0.85 and 0.81; Figure 2 and Figure 3). Few additional events occurred between one and two years of follow-up. Two TVRs occurred in the PROMUS Element cohort (a TLR and non-TLR TVR both treated with PCI). In the SYNERGY arm, two subjects died - one a cardiac death and one a non-cardiac death. In the SYNERGY ½ dose group, there were one cardiac and two non-cardiac deaths as well as one non-TLR TVR treated with PCI. The two-year rates of the combined endpoint of all-cause death, MI and TVR were similar between groups (Figure 2). All-cause death was significantly increased in the SYNERGY arm of the trial and was driven by non-cardiac causes (Table 2). The rate of cardiac death was not significantly different between randomised arms. The two cardiac deaths occurred 472 and 687 days after the index procedure; both patients died of an unknown cause which is considered a cardiac death unless proven otherwise (Online Table 1). The rates of other clinical events were similar between groups including ST (Figure 3 and Table 2). The rates of DAPT therapy were similar in all three randomised groups at two years (Online Table 2).
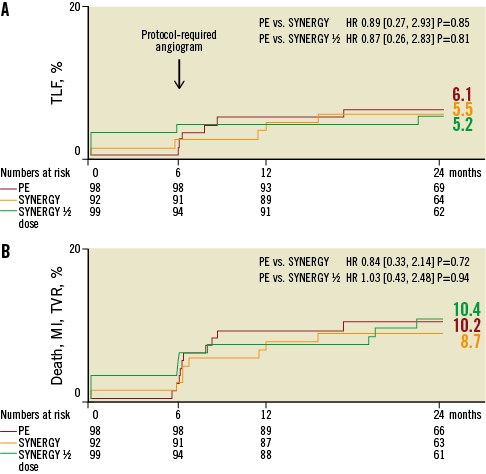
Figure 2. TLF and Death/MI/TVR through two years. Kaplan Meier cumulative event curves at two-year follow-up.
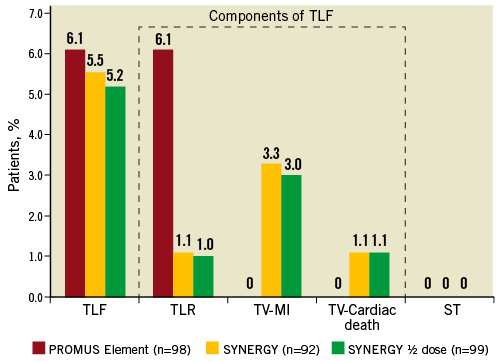
Figure 3. TLF, TLF components and ST through two years. No significant differences for two-year TLF, the individual components of TLF or ST between the arms of the EVOLVE trial were found.
Randomisation in the EVOLVE FHU trial was stratified by the presence or absence of medically-treated diabetes. TLF occurred in 4.5% (1/22) of medically-treated patients in the PE arm, 6.3% (1/16) of SYNERGY-treated diabetic patients and 5.9% (1/17) SYNERGY ½ dose diabetic patients. All cardiac events in the medically-treated diabetic patients occurred within the first year. There were no significant differences between treatment groups and the rates appeared to be similar with respect to the rest of the study population (non-medically treated diabetic patients). TLF occurred in 4.5% (1/22), 6.3% (1/16) and 5.9% (1/17) of medically-treated diabetics in the PE, SYNERGY and SYNERGY ½ dose arms, respectively. TLR and TVR were significantly increased in PE versus SYNERGY ½ non-medically treated diabetic dose patients (TVR 10.5% vs. 2.6%, p=0.049; TLR 6.6% vs. 0.0%, p=0.02) with a trend toward an increase in PE compared with SYNERGY non-medically treated diabetic patients (TVR 10.5% vs. 2.6%, p=0.054; TLR 6.6% vs. 1.3%, p=0.10).
Discussion
This study found that everolimus delivered from an ultrathin bioabsorbable abluminal polymer resulted in equivalent net volume obstruction and ISA compared with a permanent polymer everolimus-eluting stent at six months. Clinically, after two years of follow-up, no significant differences were found between the PE and SYNERGY or SYNERGY ½ dose groups for TLF, cardiac death, MI, or repeat revascularisation. One presumed cardiac death occurred in each of the SYNERGY and SYNERGY ½ dose arms of the trial; although both were due to an unknown cause. No patient in any arm experienced a Q-wave MI or ST. In comparison to PE, outcomes in the SYNERGY ½ dose arm were similar to the SYNERGY arm.
The EVOLVE trial was not powered to detect differences in TLF or other clinical outcomes between the arms of the trial at two years; thus, the results reported here should be considered hypothesis-generating only.
The results presented here are commensurate with previously reported studies comparing DES with biodegradable and permanent polymers. In several unrestricted, all-comers analyses comparing drug-eluting biodegradable polymer stents with sirolimus-eluting stents (SES), (including the LEADERS, ISAR-TEST 3 and 4 trials, as well as SORT OUT V and COMPARE II), the composite of cardiac death, MI and clinically-indicated TLR occurred in 4.6% to 12.3% of patients receiving bioabsorbable polymer-coated stents at one-year (the rate in SORT OUT V, 5.4%, included definite ST at a rate of 0.7%)18-20. Though not directly comparable due to less patient complexity (fewer patients with prior CABG, multivessel disease or chronic total occlusions) in EVOLVE, the composite event rate (target vessel-related cardiac death, target vessel-related MI and TLR) was 4.4% and 4.2% of patients in the SYNERGY and SYNERGY ½ dose arms. At two years, the rate of cardiac death, MI and clinically-indicated TLR was 11.9% versus 13.6% in a trial of BES versus SES compared with 5.5%, 5.2% and 6.1% in the SYNERGY, SYNERGY ½ dose and PE arms of EVOLVE20,21. Longer-term analyses have shown similar or better clinical outcomes when comparing biodegradable and durable polymer stents20-22.
The greatest potential advantage of a bioabsorbable drug delivery polymer on a DES (BP-DES) is that its dissolution leaves a BMS, reducing long-term durable polymer exposure and thereby potentially reducing the likelihood of late ST, even if dual antiplatelet therapy is ceased before the currently recommended six to 12 months23,24. BP-DES may have lower rates of late ST and, potentially, repeat revascularisation compared to durable polymer DES19,20,25, 26-32. The SYNERGY stent improves on existing BP-DES and durable polymer DES technology in a number of ways including a thinner and more flexible platform, shorter polymer resorption time and reduced initial polymer load. Comparatively, there is a range of both drug elution and polymer degradation rates within currently available BP-DES such that antiproliferative drug release may be complete less than three months or take up to a year and polymer degradation occurs between six months and three years19,26-32.The rapid dissolution of the polymer may mitigate the presence of the polymer as a risk factor for late ST and may increase the likelihood that DAPT could be discontinued23,24. This would be important in patients who are unable to take DAPT due to the risk of bleeding, economic reasons, allergies, resistance to antiplatelet inhibition and in the case where other medications, like Coumadin derivatives, are required concurrently. However, further study and longer-term follow-up is warranted.
Study limitations
Although this study provides important information about the safety and performance of the SYNERGY stent, there are a number of limitations. The study only included patients with relatively simple de novo lesions such that patients with acute MI, total occlusion, bifurcation, left main coronary artery and graft lesions, ostial lesions, or lesions with thrombus or excessive tortuosity or angulation were excluded. Additionally, the study was not powered to detect differences in clinical event rates or to assess the risk of thrombosis or the required duration of dual antiplatelet therapy with SYNERGY. However, though underpowered, the study still provides valuable estimates of treatment effects which can be built upon with future trials including EVOLVE II which compares the SYNERGY stent to PROMUS Element Plus. IVUS was utilised post-procedure and at six months. Given the time course of drug elution and polymer degradation, an additional imaging time point may have revealed differences between durable and degradable polymer coated stents; however, only clinical follow-up was performed in this study after six months. Finally, a two-year follow-up period may not be sufficient to reflect the true long-term outcomes in the different stent groups.
Conclusions
IVUS outcomes in patients receiving everolimus delivered from an ultrathin bioabsorbable abluminal polymer coated DES demonstrate that the antirestenotic efficacy of SYNERGY is maintained even with disappearance of the coating after the drug is eluted by 90 days. The clinical results suggest that the stent is safe and effective through two years.
Acknowledgements
The authors thank Kristine Roy, PhD (Boston Scientific Corporation) for assistance in manuscript preparation and Jian Huang, MS, MD (Boston Scientific Corporation) for statistical analysis.
Funding
This study was supported by Boston Scientific Corporation.
Conflict of interest statement
I. Meredith and N. West have received honoraria for speaking/consultancy for Boston Scientific; N. Weissman and D. Walters have received grant support from Boston Scientific; A. Banning has received unrestricted research funding from Boston Scientific and honoraria for speaking from Medtronic and Abbott Vascular; and D. Allocco and K. Dawkins are employees and stockholders of Boston Scientific Corporation. The other authors have no conflicts of interest to declare.
Online data supplement
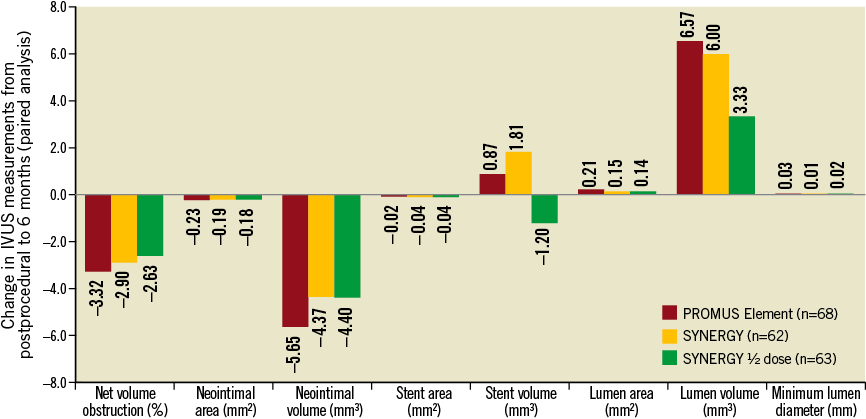
Online Figure 1. Change in intravascular ultrasound assessment from post-procedure to six months. Matched analysis of area and volume changes at the in-stent segment for the PROMUS Element (n=68 pairs), SYNERGY (n=62 pairs) and SYNERGY ½ dose (n=63 pairs) arms of the EVOLVE trial between post-procedure examination and six-months