Abstract
Aims: The combined prophylactic strategy of sodium bicarbonate plus N-acetylsyteine (NAC) seems to be effective in preventing contrast induced acute kidney injury (CI-AKI) in patients at low-to-medium risk. However, in patients at high and very high risk the rate of CI-AKI is still high. In this subset of patients the anticipated advantages of the RenalGuard™ System should be investigated. The RenalGuard™ System (PLC Medical Systems, Inc., Franklin, MA, USA) is a real-time measurement and real time matched fluid replacement device designed to accommodate the RenalGuard therapy, which is based on the theory that creating and maintaining a high urine output is beneficial by allowing a quick elimination of contrast media, and, therefore, reducing its toxic effects.
Methods and results: The REMEDIAL II trial is a randomised, multicentre, investigator-sponsored trial addressing the hypothesis that the RenalGuard System is superior to the prophylaxis with sodium bicarbonate infusion plus NAC in preventing CI-AKI in high and very high risk patients. Consecutive patients with chronic kidney disease (CKD) and at high to very high risk for CI-AKI, referred to our institutions for coronary and/or peripheral procedures, will be randomly assigned to 1) prophylactic administration of sodium bicarbonate plus NAC (control group) and 2) RenalGuard System treatment (RenalGuard group). All enrolled patients must have an estimated glomerular filtration rate ≤30 ml/min/1.73 m2 and/or a contrast nephropathy risk score ≥11. In all cases iodixanol (an iso-osmolar, non-ionic contrast agent) will be administered. The primary endpoint is an increase of ≥0.3 mg/dL in the serum creatinine concentration 48 hours after the procedure.
Conclusions: The REMEDIAL II trial will give important answers on how to prevent CI-AKI in high and very high risk patients undergoing contrast media exposure.
Introduction
The REMEDIAL II trial is a randomised, multicentre, investigator-sponsored trial addressing the hypothesis that the RenalGuard System is superior to the prophylaxis with sodium bicarbonate infusion plus NAC in preventing CI-AKI in high and very high-risk patients. Consecutive patients with chronic kidney disease (CKD) and at high to very high-risk for contrast-induced acute kidney injury (CI-AKI), referred to our institutions for coronary and/or peripheral procedures, will be randomly assigned to 1) prophylactic administration of sodium bicarbonate plus N-acetylcysteine (control group) and 2) RenalGuard System treatment (RenalGuard group). All enrolled patients must have an estimated glomerular filtration rate ≤30 ml/min/1.73 m2 and/or a contrast nephropathy risk score ≥11. In all cases iodixanol (an iso-osmolar, non-ionic contrast agent) will be administered. The primary endpoint is an increase of ≥0.3 mg/dL in the serum creatinine concentration 48 hours after the procedure.
Background
Contrast-induced acute kidney injury (CI-AKI) is a powerful predictor of unfavourable early and late outcome1-3. In the last years, several studies showed the advantages of the prophylaxis by N-acetylcysteine (NAC)4,5 and sodium bicarbonate6. In the REMEDIAL trial we have demonstrated that the combined strategy of volume supplementation by sodium bicarbonate plus NAC seems to be superior to the association of normal saline plus NAC alone or plus ascorbic acid and NAC in preventing CI-AKI in patients at low to medium risk7. However, in high- and very high-risk patients the rate of CI-AKI may be still high. One theory is that creating and maintaining a high urine output is beneficial to prevent kidney damage. This high urine flow rate should allow the body to rapidly eliminate contrast, reducing its toxic effects. Data from the PRINCE study, indeed, indicate that increasing the urine flow rate (≥150 ml/h) reduces the toxic effect of contrast media8. This effect may have a crucial benefit in patients at high- and very high-risk. However, at present, a forced diuresis regime is usually achieved by high dose of furosemide, which may be deleterious9. In a small study of 18 patients, furosemide pre-treatment (1.5 mg/kg) added to an intravenous fluid protocol was associated with significantly worse renal function than was intravenous fluid alone10. Significant weight loss was observed in the patients treated with furosemide, suggesting that the potentially deleterious effect of furosemide was the result of a negative fluid balance in this subset of patients, therefore, new and promising approaches allowing a high urine flow rate without the risk of volume depletion should be investigated.
Another important aspect that needs to be addressed is the advantage of the new biomarkers for CI-AKI. Indeed, serum creatinine (sCr) suffers for several limitations11,12. Two biomarkers seems to be very promising: cystatin C (CyC) and neutrophil gelatinase-associated lipocalin (NGAL). We recently observed that 1) a CyC increase <10% at 24 hours is a reliable marker for ruling out CI-AKI and 2) a CyC increase ≥10% at 24 hours is an independent predictor of 1-year MAE3. Preliminary data suggests that urinary NGAL level significantly increases at two hours following contrast exposure, allowing a quick diagnosis of AKI.
Study hypothesis
The RenalGuard™ System (PLC Medical Systems, Inc., Franklin, MA, USA) in combination with a limited (0.25 mg/kg) dose of furosemide increases the urine flow rate. This combination (RenalGuard™ System plus furosemide) allows the achievement of a high urine output safely by maintaining the intravascular blood volume and minimising the risk of over- or under-hydration. The potential benefits of the RenalGuard therapy are intended to reduce the incidence of CI-AKI via a combination of known physiological effects of high urine output including: a) lower concentration of contrast in the kidneys, b) more rapid transit of contrast through the kidneys, c) less overall exposure to the toxic contrast agent, and d) potential reduction of oxygen consumption in the medulla.
Preliminary data suggests that the RenalGuard System allows a quick renal first-pass elimination and therefore reduces the risk for CI-AKI13,14. In the US pilot clinical trial, a 250 ml bolus of saline and a diuretic was used to create a high urine rate and the matched replacement helped maintain urine output without risk of over- or under-hydration13. Patients at high- and very high-risk for CI-AKI represent the ideal population to test whether the combination of the RenalGuard System plus a low-dose of furosemide 1) allows to reach the urine flow target ≥300 ml/h, and 2) is more effective than the combined strategy of volume supplementation by sodium bicarbonate plus NAC in preventing CI-AKI.
Methodology
Patient population
This multicentre, randomised, investigator-sponsored study will compare two different strategies to prevent CI-AKI in patients at high- to very high- risk for CI-AKI. All consecutive patients with chronic kidney disease (CKD) scheduled for coronary and/or peripheral angiography and/or angioplasty with an eGFR ≤30 ml/min/1.73 m2 and/or a risk score ≥11 will be considered eligible for this study (Figure 1). Estimated glomerular filtration rate (eGFR) will be calculated by applying the Levey modified Modification of Diet in Renal Disease formula: (186.3×serum creatinine –1.154)×(age–0.203)×(0.742 if female)15. CKD is defined as an estimated glomerular filtration rate (eGFR) <60 ml/min/1.73 m2,15. The risk score for predicting CI-AKI is reported in the Table 1.
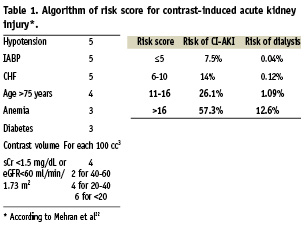
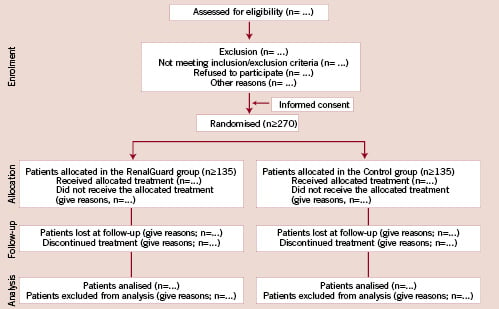
Figure 1. Diagram showing the flow of participants through each stage of the trial according to the CONSORT guidelines.
Recruitment, enrolment and allocation
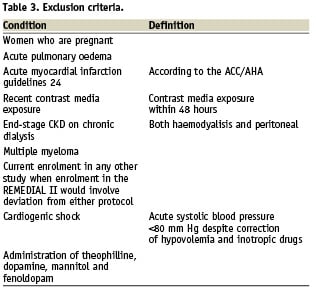
All patients with CKD scheduled for coronary and/or peripheral angiography/angioplasty from January 2009 will be screened for inclusion/exclusion criteria (Figure 1; Tables 2 and 3). All patients with inclusion/exclusion criteria satisfied and who will sign the informed consent will be enrolled into the study. We expect that approximately 10% of the global population treated will satisfy the inclusion/exclusion criteria. Therefore the estimated overall study duration is approximately one year. The REMEDIAL II trial will be conducted in across four interventional cardiology centres in Italy, according to the principles of the Declaration of Helsinki16 and Good Clinical Practice17 and has been approved by our Ethic Committees. The trial is registered with www.clinicaltrial.gov (trial ID NCT01098032). The number of patients screened, randomised, treated and analysed will be reported according to the CONSORT guidelines (Figure 1).

Protocol
Following enrolment, patients will be randomly assigned to one of the following treatments: 1) Control group: intravenous sodium bicarbonate plus NAC administration, and 2) RenalGuard group: prophylactic controlled hydration with saline (0.9%) plus NAC (1500 mg in one litre of saline) (Figure 1). Both these therapies will be instituted before and after administration of the contrast agent. In all patients the left ventricular end-diastolic pressure will be measured by a pigtail catheter at the beginning of the procedure.
Control group
Patients allocated to the control group will receive 154 mEq/l of sodium bicarbonate in dextrose and H2O, according to the protocol reported by Merten et al.6 The initial intravenous bolus is 3 ml/kg per hour for one hour immediately before the contrast injection. Following this, patients will receive the same fluid at a rate of 1 ml/kg per hour during contrast exposure and for six hours after the procedure. All patients will receive NAC (Fluimucil; Zambon Group SpA, Milan, Italy) orally at a dose of 1,200 mg twice daily on the day before and on the day of administration of the contrast agent (total of two days)18. An additional NAC dose (at least 1,200 mg) will be intravenously administered during the procedure. The total NAC dose is at least 6 g.
RenalGuard group
Patients enrolled in the RenalGuard group will be treated by hydration with saline (0.9%) plus NAC (1500 mg in one litre of saline) controlled by the RenalGuard system (Figure 2). This system includes:
1) closed loop fluid management system
2) high volume fluid pump
3) high accuracy dual weight measuring system
4) motion detection artefact reduction
5) proprietary: single use intravenous set; urine collection system that interfaces with a standard Foley catheter
6) real-time display of urine and hydration fluid volume
7) user set net fluid gain and fluid bolus administration for patients that require re-hydration prior to and during the procedure
8) automatic battery back-up
9) automatic priming of infusion set
10) timely alerts to drain urine bag or to replace the hydration fluid bag
11) safely features such as automatic air detection and detection of IV occlusions.
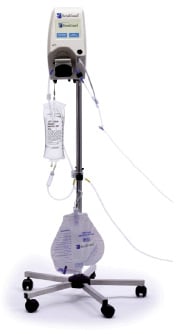
Figure 2. The RenalGuard system.
In the RenalGuard group, an initial bolus (priming) of 250 ml will be administered (Figure 3). In case of left ventricular dysfunction (ejection fraction ≤30% as assessed by 2D-echocargiography) and/or unstable haemodynamic conditions (recent [<7 days] pulmonary oedema or acute heart failure) the bolus will be reduced to ≤150 ml. Following the initial bolus, furosemide (0.25 mg/kg) will be administered in order to achieve the optimal urine flow (≥300 ml/h). As soon as the urine flow reaches the target value, the patient will be moved in the catheterisation laboratory and the procedure will start. The controlled hydration by the RenalGuard system will be continued during the procedure and for four hours following the procedure. Urine flow is monitored and maintained at the target value through the procedure and during the following four hours. Additional furosemide doses are allowed in case of a decrease of the urine flow below the target value. A nurse will record the values (total infusion and urine volumes, and urine flow rate) every 15 minutes on a dedicated form, see online appendix.
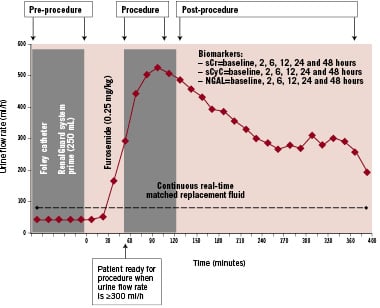
Figure 3. Diagram showing each stage of the RenalGuard therapy.
Biomarkers of kidney function and injury
Serum creatinine, cystatin C, blood urea nitrogen, sodium and potassium will be measured the day before, 2, 6, 12, 24, 48 hours and one week after administration of the contrast agent; additional measurements will be performed in all cases of deterioration of the baseline renal function. In patients randomised in the control group urinary pH will be measured at the time of randomisation and during the treatment (that is, after infusion of the bolus when the patient spontaneously voided). Urine and plasma (NGAL) will be measured the day before, 2, 6, 12, 24 and 48 hours after contrast media exposure.
Urine will be collected in all patients at baseline, at six and 24 hours after the contrast exposure. The sediment obtained by centrifugation at 1500 rpm for 10 minutes is aliquoted to prepare both cyto-spins and cell-blocks (CB). The presence of tubular cells is assessed by standard Papanicolaou staining and by galectin-3 staining. CBs are employed to stain tubular cells for their marker Gal-3; as a marker of cell damage caspase-3, p53 and p21WAF1 are adopted.
Contrast agents
Iodixanol (Visipaque®; [GE Healthcare Ltd, Little Chalfont, Buckinghamshire, United Kingdom], a non-ionic, iso-osmolar [290 mOsm/kg of water]) contrast agent will be used in all patients.
Study endpoints
The primary outcome measure is development of CI-AKI, defined as an increase in the serum creatinine concentration ≥0.3 mg/dL from the baseline value at 48 hours after the administration of the contrast media or the need for dialysis19. This is the Acute Kidney Injury Network (AKIN) criteria for acute kidney injury. Definitions of AKI include both absolute and percentage increases in sCr20. As recently demonstrated also in an experimental model, an absolute increase in sCr seems to be better suited for the early diagnosis of acute kidney injury than a percentage increase21. Secondary endpoints are 1) an increase in the serum creatinine concentration ≥0.25% and ≥0.5 mg/dl at 48 hours after contrast exposure; 2) changes in the serum cystatin C concentration at 24 and 48 hours after contrast exposure, 3) changes in the urine and serum NGAL concentration after contrast exposure, 4) the rate of acute renal failure requiring dialysis (defined as a decrease in renal function necessitating acute haemodialysis, ultrafiltration or peritoneal dialysis within the first five days post-intervention), 4) the rate of in-hospital, 6- and 12-month major adverse events, and 5) the changes in urine tubular cells number and presence of marker of cell damage after contrast media exposure. Major adverse events (MAE) will be considered: a) death, b) renal failure requiring dialysis, and c) acute pulmonary oedema.
Data collection and monitoring
Patient demographic details, medical history, current medication, eGFR, risk score for CI-AKI, and left ventricular ejection fraction are recorded at baseline. Total hydration volume administered according to the prophylaxis and during the 24 and 48 hours following the procedure as well as the total urine volume will be recorded. The pre-procedure sCr level is considered as the SCr concentration before the initiation of any prophylaxis. Endpoint data and adverse events are collected during the in-hospital stay. Six-month and 12-month MAE will be also collected.
All adverse events are recorded in the case report form and the data coordinating centre will be informed by facsimile within 72 hours of any events. Serious events and any other safety issues will be reviewed by an independent Data Monitoring and Safety Committee. All events will be adjudicated by a Clinical Events Committee (CEC), who will be blinded to the treatment assignment. At least two members of the CEC will review clinical data and relevant documentation and determine whether endpoints have occurred according to the study definitions. In case of disagreement between the reviewers, a third member of the CEC will adjudicate and the data will be considered by the entire committee if two of the three reviewers do not agree.
Statistical analysis
Treatment assignment between the two groups will be determined by randomisation in a 1:1 ratio. To ensure that almost equal number of patients receive one of the two treatments, randomisation blocks of four will be used (Plan Procedure of SAS, version 8.2; SAS Institute Inc., Cary, NC, USA). The sample size was selected to demonstrate a reduction in the primary endpoint of CI-AKI from 25% in the control group to 10% in the RenalGuard group. Using a two-sided Chi-square test with a significance level of 0.05, a total of at least 270 randomised patients (135 in each arm) afforded the study 90% power. The expected CI-AKI rate in the Control group has been based on the published data1,3,7,22,23. According to Mehran et al22, the expected CI-AKI rate in patients with a risk score 11-16 and ≥16 is 26.1% and 57.3%, respectively. In the study by Marenzi et al23 enrolling patients with eGFR <30 ml/min/1.73 m2 the CI-AKI in the control group was 50%. We recently observed that in patients with eGFR <30 ml/min/1.73 m2 pre-treated with sodium bicarbonate plus NAC the rate of CI-AKI (defined as an increase in the serum creatinine concentration ≥0.3 mg/dL from the baseline value at 48 hours) is approximately 29% and in patients with risk score ≥11 is 21%3.
Appendix
Steering committee: Carlo Briguori, Gabriella Visconti, Bruno Ricciardelli, Gerolama Condorelli.
Data Monitoring and Safety Committee: Flavio Airoldi (Chairperson), Francesca De Micco, Amelia Focaccio, Rosalia Giannone
Clinical Events Commitee: Bruno Golia, Cristina Quintavalle, Gianluca Caiazzo
Clinical Trials and Evaluation Unit Members: Carlo Briguori (Project manager), Ciro Zanca, Margherita Iaboni, Natalia V. Rivera (statistician).
REMEDIAL II Centers
Clinica Mediterranea, Naples; Investigators: Carlo Briguori, Gabriella Visconti, Amelia Focaccio; Study Coordinator: Francesca De Micco
IRCCS Multimedica, Milan; Investigators: Flavio Airoldi, Davide Tavano, Silvio Bertoli; Study Coordinator: Tiziana Staine
University of Ferrara, Ferrara; Investigators:Marco Valgimigli, Roberto Ferrari; Study Coordinator: Monia Monti
University of Modena, Modena; Investigators: Giuseppe Massimo Sangiorgi, Mariagrazia Modena; Study Coordinator: Simona Lambertini
