Abstract
Aims: This prospective, non-randomised, observational study conducted in Europe was designed in order to assess the long-term safety and efficacy of the Parachute device in ischaemic heart failure subjects as a result of left ventricle remodelling after anterior wall myocardial infarction.
Methods and results: One hundred subjects with New York Heart Association Class II-IV ischaemic heart failure (HF), ejection fraction (EF) between 15% and 40%, and dilated akinetic or dyskinetic anterior-apical wall without the need to be revascularised were enrolled. The primary safety endpoint was procedural- or device-related major adverse cardiac cerebral events (MACCE). The secondary safety endpoint was the composite of mortality and morbidity. Secondary efficacy endpoints included haemodynamic measurements determined by echocardiography, LV volume indices, and assessment of functional improvement measured by a standardised six-minute walk test. Of the 100 subjects enrolled, device implantation was successful in 97 (97%) subjects. The one-year rates of the primary and secondary safety endpoints were 7% and 32.3%, respectively. The secondary endpoints, LV volume reduction (p<0.0001) and six-minute walk distance improvement (p<0.01), were achieved.
Conclusions: The favourable outcomes observed in this high-risk population provide reassuring safety and efficacy data to support adoption of this technology as a therapeutic option for HF subjects. Clinical registration: www.clinicaltrials.gov numbers are NCT01297296 (PARACHUTE III) and NCT01286116 (cohort B subjects).
Introduction
Left ventricle (LV) dilation and remodelling after acute myocardial infarction have been well characterised in experimental and clinical investigations1,2. Progressive LV dilation and remodelling post myocardial infarction occurred in 33% of subjects enrolled in the GISSI-3 trial (n=13,679)3. Symptomatic post-MI LV dilation is more common in subjects with an anterior infarction4. With current aggressive invasive strategies and intensive pharmacotherapy, acute myocardial infarction (AMI) survival rates have increased, but may have created a larger population with greater residual myocardial injury and a higher risk of developing heart failure (HF)5. Once hospitalised for HF, the overall one-year mortality remains unacceptably high at 32%, in spite of modern pharmacological and mechanical approaches5. Therapeutic efficacy depends, albeit not exclusively, on improvements in LV volumes, reduced wall stress and restored geometry, since left ventricular end-systolic and end-diastolic volumes are surrogate measures of LV remodelling and independent clinical predictors of outcomes in HF subjects6.
The concept of percutaneous ventricular restoration (PVR) of the LV is based on the premise that a dedicated partitioning, compliant device delivered via a catheter-based approach may achieve LV volume reduction, geometric reconfiguration, and synchronised wall motion to achieve a more effective ejection while minimising the risk of a surgical approach7. Initial studies8,9 in subjects with post-MI remodelling and symptoms of New York Heart Association (NYHA) Class II-IV HF demonstrated safety, feasibility, and favourable LV function as measured by LVESVi, LVEDVi, EF and LVEDP. Moreover, improved symptoms and functional status were confirmed after 12 months. In these early PVR studies, clinical outcomes of mortality, hospitalisations for heart failure, and stroke were also improved compared to literature reports of devices in a similar population10-14 where functional outcomes and/or hospitalisation for HF were improved. Three-year outcome results from the PARACHUTE first-in-human study in post-MI subjects with NYHA Class II-IV heart failure were recently reported15.
Methods
STUDY DESIGN
This prospective, non-randomised, observational study conducted in 20 medical centres across 10 countries in Europe was designed to assess the long-term safety and efficacy of the Parachute device (CardioKinetix, Inc., Menlo Park, CA, USA). The study combined enrolment from two contemporaneous studies with consecutive enrolment and the same inclusion and exclusion criteria, Europe cohort B and PARACHUTE III, which will be collectively referred to as PARACHUTE III. Europe cohort A was the first-in-man cohort enrolled four years prior to cohort B and was not included in this analysis for that reason. Following implantation of the device, clinical and echocardiographic follow-up was performed at six months and one year (planned annually thereafter up to five years).
SUBJECT SELECTION
The study included subjects with symptomatic ischaemic HF of NYHA Class III, IV, or Class II if diagnosed with NYHA Class III or IV during a three-month period prior to enrolment. The subjects were at least 18 years of age with LV wall motion abnormalities (anteroapical akinesis or dyskinesis) secondary to MI, an LV ejection fraction between 15% and 40%, and managed with stable doses of standard HF medical therapy for at least three months, as determined by the investigator. Subjects with myocardial ischaemia who underwent revascularisation or cardiac resynchronisation therapy within 60 days of enrolment and those with significant valve disease were excluded from the study. All sites obtained approval from an ethics committee before study commencement, and written informed consent was obtained for all subjects at the appropriate time prior to involvement in the study.
STUDY DEVICE AND PROCEDURE
The Parachute system includes the device (Figure 1), a delivery system with a balloon that facilitates expansion of the device, and a pre-shaped delivery catheter and dilator. The Parachute device comprises a self-expanding nitinol frame, an ePTFE impermeable membrane, and an atraumatic polymer foot, which has eight sizes (65, 75, 85 and 95 mm diameter, each offered in two “foot” heights). The distal atraumatic foot is radiopaque and provides a contact point between the LV apical wall. The contact point is selected in order to orient the device with a vector towards the outflow tract.
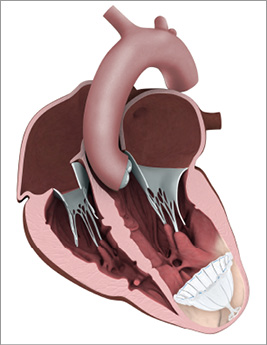
Figure 1. Illustration of the Parachute device implanted in a dilated left ventricle.
The procedure is illustrated in Figure 2 and is performed in a catheterisation laboratory with the subjects usually under conscious sedation15. Subjects were considered intention-to-treat if i) they signed a consent form, ii) they underwent a baseline evaluation, and iii) the Parachute procedure was attempted by passage of the implant past the guide catheter. Device size selection was based on multislice computed tomography which also allowed identification of any LV apical thrombus, pseudo-chordae, or severe calcification, which could have precluded safe deployment. All subjects received low-dose aspirin and anticoagulation with warfarin for at least 12 months post device implant.
![]()
Figure 2. Sequence of a Parachute implantation in the left ventricle (LV). A) Pigtail in the LV cavity to perform LV angiography. B) Device placement with foot exposed and in contact with the antero-apical wall. C) Balloon inflation to facilitate self-expansion of the device. D) Device fully expanded but still attached to the delivery system. E) Final positioning after release of the device.
DATA COLLECTION AND OVERSIGHT
All study-related data were collected on standardised electronic case report forms. All study-mandated echocardiograms were sent to an independent core laboratory (Yale Cardiovascular Research Group, Yale University School of Medicine, New Haven, CT, USA). Data management was performed by an independent contract research organisation. An independent clinical events committee (CEC) adjudicated serious adverse events (SAEs), cardiac and non-cardiac mortality, and hospitalisations for HF related to either device or procedure.
STUDY ENDPOINTS
The primary endpoint of the PARACHUTE III study was the assessment of long-term safety as measured by procedural- or device-related major adverse cardiac cerebral events (MACCE) in real-world use of the Parachute device up to five years post procedure; one-year safety data are reported here. MACCE was broadly defined and included the occurrence of any of the following: death, cardiac death, MI, emergent cardiac surgery, erosion of the device through the LV, cardiac tamponade, peripheral embolisation (including stroke), new or worsening HF, endocarditis or device infection, device migration or embolisation, or placement of a mechanical support device. Worsening HF was defined as an unplanned hospitalisation that resulted in at least one overnight stay (i.e., where the admission date and the discharge date were different) that included increased signs and/or symptoms of worsening HF including increased jugular venous pressure (JVP) and required the administration or augmentation of intravenous HF therapy (e.g., inotropes, diuretics, and/or vasodilators). Vascular complications were defined using an expanded Valve Academic Research Consortium (VARC) definition which even included damage of the aortic valve requiring surgery16. The secondary safety endpoint was the composite of mortality and morbidity. Secondary efficacy endpoints included haemodynamic measurements determined by echocardiography, LV volume indices, and assessment of functional improvement measured by a standardised six-minute walk test (6MWT). A clinically meaningful change in the six-minute walk test was defined as an absolute change of 20 metres.
Statistical analysis
The primary endpoint and secondary safety endpoint analysis will be performed on the intention-to-treat population. Separate analysis for the effectiveness endpoints will be performed on the as-treated population. Baseline characteristics were summarised using mean±SD for continuous variables and counts and percentages for categorical variables. All statistical comparisons were two-sided t-tests or paired t-tests with alpha=0.05. MACCE over time was evaluated using Kaplan-Meier analysis. All analyses were performed using SAS version 9.3 (SAS, Cary, NC, USA).
Results
SUBJECT POPULATION
Between June 2011 and May 2013, 100 subjects (Online Appendix, Table 1) were enrolled into the study. Of the 100 subjects, 55 came from the Europe cohort B study and 45 from the PARACHUTE III study. The protocol inclusion and exclusion criteria were the same for both studies and all subjects selected from both studies are included. Ninety-seven subjects were treated with the Parachute device and followed up for haemodynamic, functional, and clinical outcomes. The disposition of subjects who completed one-year follow-up is shown in Figure 3.
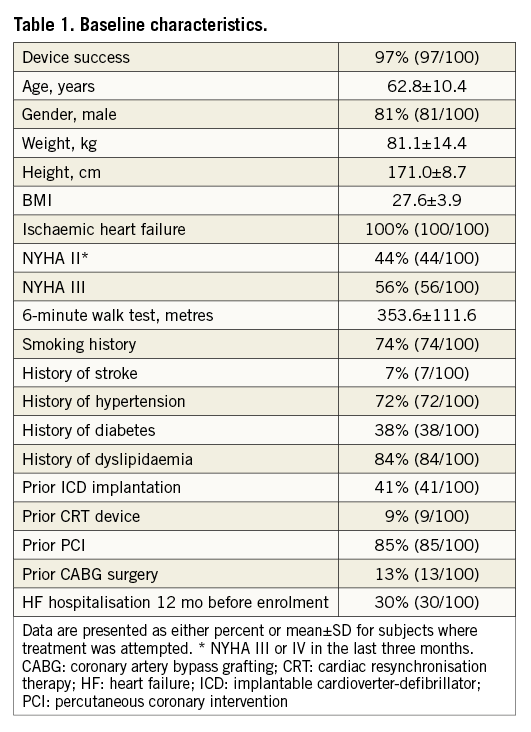
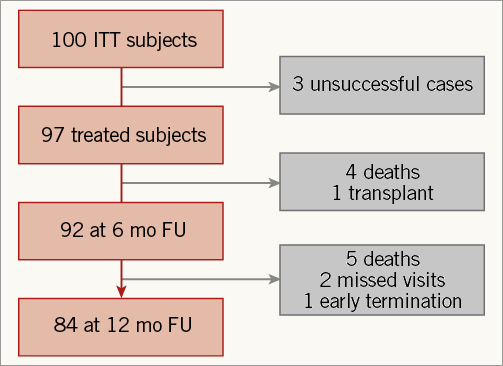
Figure 3. Disposition of intention-to-treat subjects. The three unsuccessful cases were due to non-optimal positioning of a device resulting in a snared removal (1), and LV perforation by the guide catheter requiring surgery (2).
BASELINE CHARACTERISTICS
While all of the subjects were required to have had a diagnosis of NYHA Class III or IV during the three-month period prior to enrolment to be included in the study, 56% were Class III and 44% were Class II at the time of their baseline assessment. Ischaemic heart disease was present in all subjects, and approximately one third had been hospitalised for HF in the previous 12 months. Most subjects were male (81%), and 85% had a prior percutaneous coronary intervention (PCI). Nearly all subjects were on a beta-blocker (97%), with 79% taking a beta-blocker, diuretic, and an ACE or ARB.
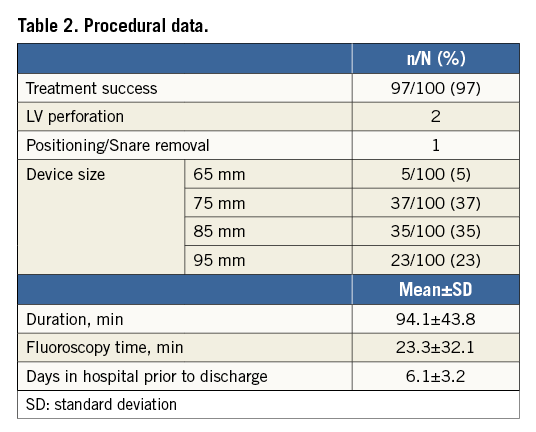
PROCEDURAL OUTCOMES AND COMPLICATIONS
Of the 100 intention-to-treat subjects, the procedural success rate was 97% (Table 2). In three subjects the device was explanted prior to discharge: one for non-optimal positioning of a device resulting in a snared removal and two for LV perforation by the guide catheter, requiring surgery. Major vascular complications were noted in 11% (11/100) of subjects, consisting of four life-threatening or disabling bleeding events due to an overt source of bleeding requiring more than four units of blood, three aortic valves requiring surgical repair, two LV perforations preventing successful implantations, one bradycardia due to AV block prior to the Parachute deployment that was considered life-threatening, and one case of mitral apparatus damage. Minor vascular complications were infrequent, related to the femoral access site, and were noted in 4% (4/100) of subjects.
MEDICAL THERAPY
The medical therapy was consistent from inclusion into the trial up to one-year follow-up for the subjects. The proportion of subjects on the following medication at baseline and one-year follow-up, respectively, was ACEi 68% versus 69%, ARB 25% versus 24%, beta-blockers 97% versus 94%, diuretics 86% versus 91%, and anti-arrhythmic 18% versus 17%. The changes in proportions of medications taken were not statistically significant. The proportion of subjects taking a beta-blocker, diuretic, and an ACE or ARB at one year remained unchanged as compared to baseline.
LONG-TERM SAFETY OUTCOMES (MACCE)
The primary safety endpoint (MACCE) at one year was 7.0% and was used to assess the long-term safety as measured by procedural- or device-related MACCE (Table 3, Figure 4). All seven of the events occurred no later than 10 days after the procedure. There was a total of 93 (93%) subjects without a device-related major adverse cardiac or cerebral event. The most frequently reported MACCE was emergent cardiac surgery. There were no device dislodgements or pericardial effusions requiring clinical intervention caused by the device. Additionally, as assessed by the echo core lab, 3.3% of subjects had thrombus on the device at one year all of which were without peripheral embolisation.
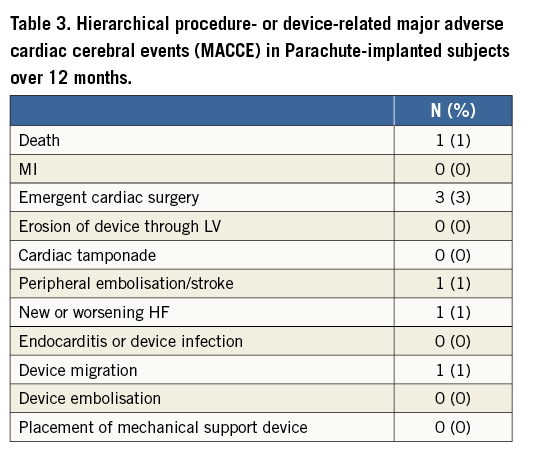
Figure 4. Primary endpoint Kaplan-Meier plot.
HAEMODYNAMICS AND FUNCTIONAL OUTCOMES
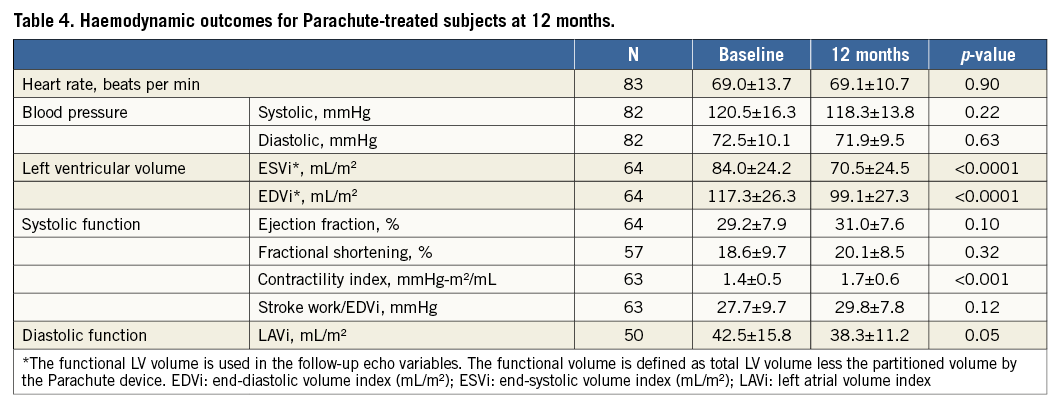
The secondary endpoints, LV volume reduction (p<0.0001) and six-minute walk distance improvement (p<0.01), were achieved with statistical significance. Improvement of systolic cardiac function was noted in LV volume indices and contractility index, along with trends in ejection fraction and stroke work index, at one-year follow-up relative to baseline values. This was accompanied by a significant reduction (p=0.05) in left atrial volume (Table 4).
Moreover, the mean NYHA class of subjects (2.0±0.7) was significantly reduced after one year (p<0.001) from baseline NYHA class (2.6±0.5), reflecting functional improvement. The proportion of subjects who improved at least one NYHA class was 43%, while 37% had no change in their NYHA class (Table 5).
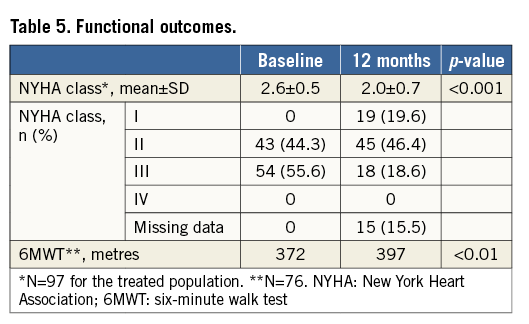
Performance on 6MWT was also improved from 372 metres at baseline to 397 at 12 months (p<0.01) (Table 5), with 46% of subjects improving by 20 metres or more compared to baseline.
CLINICAL OUTCOMES
The composite secondary endpoint rate of mortality and morbidity was 32.3% and included all-cause death (10), hospitalisation for HF (12), myocardial infarction (9), and stroke (3) (Table 6, Figure 5). The average time to first event was 113±98 days. The more contemporary endpoints utilised in HF trials of mortality and the combination of mortality and HF hospitalisation had one-year results of 9.5% and 26.0%, respectively.
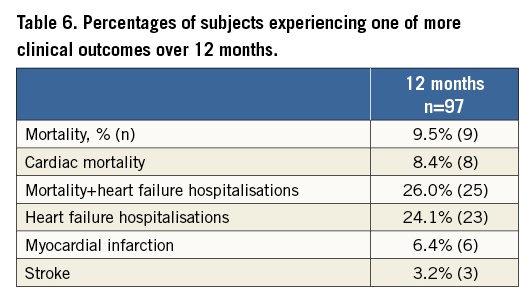
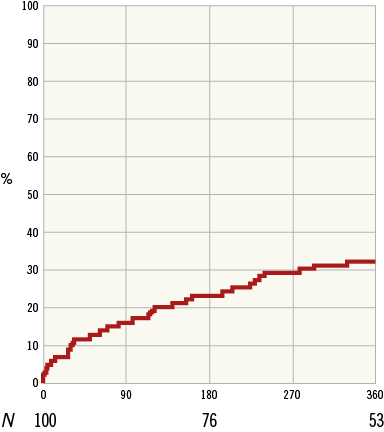
Figure 5. Secondary endpoint (mortality and morbidity) Kaplan-Meier plot.
Discussion
The PARACHUTE III study confirms the safety of PVR therapy to treat subjects with ischaemic HF and akinetic or dyskinetic anterior-apical wall. The outcomes observed in subjects treated with the Parachute device demonstrated the safety and efficacy of PVR in a real-world setting. The data were consistent with previously reported results15. These results showed a significant reduction of left ventricle and left atrial volumes for up to one year post implant, and concomitant improvements in HF symptoms. Most notable from a subject perspective is that nearly half demonstrated clinically significant improvement in the 6MWT. It has been well documented that functional capacity of HF subjects declines over time6. Against this declining profile of HF subjects, approximately 75-80% of the Parachute population is improving (approximately 50%) or maintaining (approximately 30%) functional status at one year.
Product development efforts have been implemented to address the potential for aortic valve damage during initial crossing into the LV. The design change is to taper the dilator to create a smooth transition from the guide catheter down to a 0.035” wire.
A recent meta-analysis using mortality data from 30 trials with a median follow-up of 17 months highlighted the critical role of LV end-systolic and end-diastolic volume reduction for the success of pharmacological and mechanical approaches to HF17. A curve from this publication correlates the amount of end-systolic volume (ESV) reduction with the predicted probability of showing a reduction in mortality. A 25 mL reduction of ESV corresponds to a predicted probability of a mortality reduction of more than 80%. The ESV as reduced by the Parachute device was sustained for one year, ranging between 30 mL and 40 mL. The Parachute data reported must be interpreted with caution as the compiled data did not include mechanical or direct reduction of LV volume. Nevertheless, the current low mortality rate of 9.5% in this population does hold promise that mechanically reducing ESV can impact on mortality rates in an HF population.
The unique Parachute mechanism of action combines the concepts and principles from the surgical ventricle restoration procedure popularised by Dor, Menicanti, and Suma18 and distinctive features of a mechanical device made of nitinol to be delivered percutaneously under fluoroscopy. The Parachute device is the first device designed specifically for PVR to treat subjects with ischaemic cardiomyopathy akinetic/dyskinetic anterior wall motion and HF symptoms. The device shape was designed to partition the damaged, non-contractile myocardium while creating a new apex and restoring the elliptical shape of the LV cavity in a reproducible manner. The device also permits contraction of any underlying healthy myocardium. The percutaneous nature of the procedure, which is performed under local anaesthesia, minimises operative risk and avoids suture-related myocardial scar. There are several possible mechanisms by which the implant of the Parachute device may improve cardiac performance and explain these initial positive results. The first is that reduction of volumes and partition of non-viable myocardium may reduce contractile wall stress. The second is that the shape of the device and its nitinol framing may help to restore the elliptical shape of the LV cavity and allow systolic torsion, respectively. The third is the substitution of the scarred, eccentric anteroapical wall motion with a compliant device that promotes synchronised contraction and actively aids in diastolic relaxation with outward radial force using stored kinetic energy, much like a trampoline19. The benefits of synchronised apical wall motion throughout the cardiac cycle allow improved cardiac output and reduced filling pressure.
In order to match the six-month cardiac function of a Parachute subject with a computational model, the material properties of the model had to be adjusted20. The principal finding was that the six-month model was associated with a decrease in LV diastolic stiffness and a large reduction in myofibre stress at end diastole. Another element supporting the fact that the Parachute affects diastolic compliance is the demonstration of remodelling of the left atrium, which is a surrogate measure for reducing left ventricle end-diastolic filling pressure. Cardiac resynchronisation therapy (CRT) is the only other medical device for HF subjects that has shown left atrial remodelling21.
In comparison to other structural heart and HF devices, the vascular complications are less than those observed in transcatheter aortic valve replacement (TAVR) which uses a similar access method, while the haemodynamic and functional improvements are similar to those observed in CRT10,21,22. The ongoing US pivotal trial, PARACHUTE IV, will enrol 560 subjects and will complement the level of evidence for this technology since it will be the first trial randomising against all appropriate medical therapy (AAMT) according to the current ACC/AHA guideline update for the diagnosis and management of chronic heart failure23.
Limitations
The study does have limitations given its unblinded, single-arm nature. Because of this study design, one cannot rule out potential bias in the adjudication process, and without a control group conclusions should be made with caution. The vascular access bleeding events were adjudicated using the original VARC definitions and not the most recent VARC-2 definitions.
Conclusions
The favourable outcomes observed in this high-risk population provide reassuring safety and efficacy data to support adoption of this technology as a therapeutic option for heart failure subjects. The ongoing large-scale, randomised clinical trial in the USA will potentially validate the present results and establish the role of this novel therapeutic approach for subjects with ischaemic HF.
| Impact on daily practice The number of subjects with HF continues to grow every year with limited therapeutic options. This report provides data on the potential of a novel technology, the Parachute device, to improve outcomes among select subjects with post-myocardial infarction heart failure. The favourable outcomes observed in this high-risk population provide reassuring safety and efficacy data to support adoption of this technology as a therapeutic option for HF subjects. |
Appendix
Enrolling centres: University of Rostock, Germany (14); Pauls Stradins Clinical University Hospital, Latvia (13); Clinical Center of Serbia, Serbia (10); Asklepios Klinik St. Georg, Germany (8); Hospital Santa Marta, Portugal (8); Ferrarotto University Hospital Catania, Italy (7); University of Heidelberg, Germany (6); Antwerp Cardiovascular Institute Middelheim, Belgium (5); Centro Hospitalar de Vila Nova de Gaia, Portugal (5); Kerckhoff Clinic, Germany (4); St. Thomas’ Hospital, United Kingdom (4); OLV Hospital Aalst, Belgium (3); Arzt St. Marien-Hospital, Germany (2); Hospital Clinico de Barcelona, Spain (2); Papworth Hospital NHS, United Kingdom (2); Golden Jubilee Hospital, United Kingdom (2); University Clinic Center Ljubljana, Slovenia (2); University College London The Heart Hospital, United Kingdom (1); University of Amsterdam Academic Medical Center, The Netherlands (1); University Medical Center Utrecht, The Netherlands (1).
Acknowledgements
Stephan Kische, MD, PhD, and Gitta Knoop, University of Rostock, Rostock, Germany (trial conduct); Mary Jo Rizzo, RDMS, RDCS Yale, New Haven, CT, USA (echo core lab); Claudia Tamburino, MD, Ferrarotto University Hospital Catania, Catania, Italy (trial conduct); Venita DePuy, PhD, Bowden Analytics, Apex, NC, USA and Clinipace Worldwide, Morrisville, NC, USA (statistics); Hiram Bezerra, MD, PhD, University Hospitals Case Medical Center, Cleveland, OH, USA (CT core lab).
Funding
The PARACHUTE III trial was funded by CardioKinetix, Inc. No extramural funding was used to support the creation of this manuscript. The authors are solely responsible for the design and conduct of this study, the study analyses, the drafting and editing of the paper and its final contents.
Conflict of interest statement
M. Thomas, H. Ince, U. Schäfer, V. Gama Ribeiro, and C. Tamburino are paid consultants for CardioKinetix. The other authors have no conflicts of interest to declare.
