Abstract
Despite significant advances in the medical management of patients living with heart failure, there continues to be significant morbidity and mortality associated with the condition. There is a growing need for research and development of additional modalities to fill the management and treatment gaps, reduce hospitalisations and improve the quality of life for patients living with heart failure. In the last decade, there has been a rapid rise in the use of non-valvular catheter-based therapies for the management of chronic heart failure to complement existing guideline-directed management. They target well-defined mechanistic and pathophysiological processes critical to the progression of heart failure including left ventricular remodelling, neurohumoral activation, and congestion. In this review, we will explore the physiology, rationale, and current stages of the clinical development of the existing procedures.
Introduction
Heart failure (HF) is a progressive syndrome characterised by maladaptive remodelling of cardiac chambers, increased wall stress and neurohumoral activation that perpetuate increased myocardial wall stress with volume overload and a risk of end-stage pump failure. There have been significant advances in the management of patients living with HF, particularly HF with reduced ejection fraction (HFrEF). This includes treatment with beta blockers1, renin-angiotensin-aldosterone system antagonists234, angiotensin receptor-neprilysin inhibitors (ARNI)5, implantable cardiac defibrillator (ICD) use6, cardiac resynchronisation therapy (CRT)7 and more recently, the use of sodium-glucose cotransporter-2 (SGLT-2) inhibitors89. Despite these improvements in the standard of care, advanced HF remains associated with significant morbidity and mortality10. In 2018, 6.2 million people were living with HF in the United States alone, and HF accounted for 13% of total mortality10. Furthermore, the costs associated with the management of HF are expected to rise to above $60 billion by 20301011. As such, there is a growing need for research and development of additional modalities to fill the management and treatment gaps, reduce hospitalisations and improve the quality of life (QOL) of people living with HF.
In the last decade, there has been a rapid rise in the use of non-valvular catheter-based therapies for the management of chronic HF to complement existing guideline-directed management. They target well-defined mechanistic and pathophysiological pathways critical in the progression of heart failure, including left ventricular (LV) remodelling, neurohumoral activation, and congestion (Central illustration).
In this review, we will explore the physiology, rationale, and current stages of the clinical development of existing procedures. The scope of this article and the devices covered are summarised in the Central illustration.
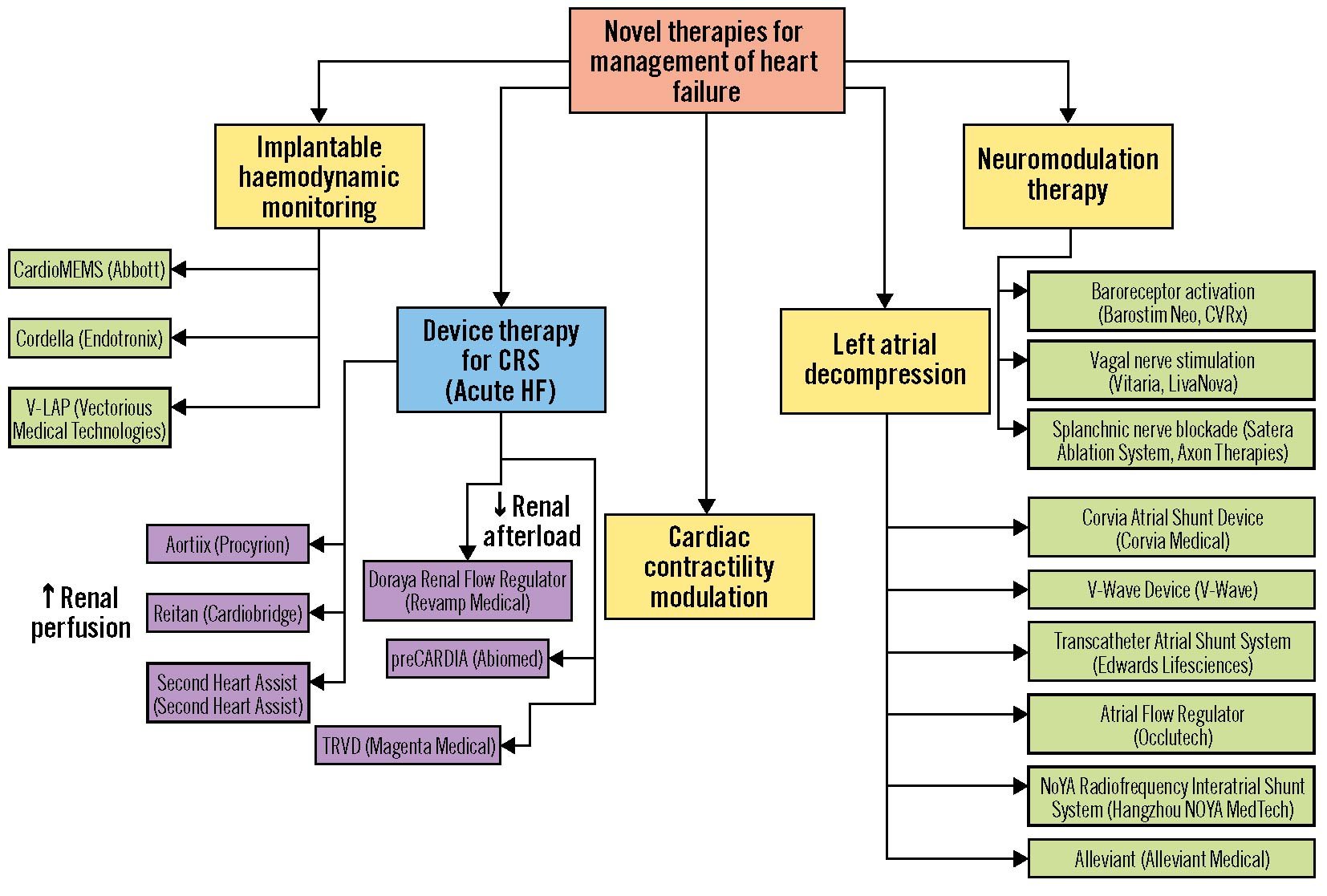
Central illustration. Novel therapies for the management of heart failure by mechanism. Yellow/green: Devices for management of chronic heart failure. Blue/purple: Devices for management of acute decompensated heart failure. CRS: cardiorenal syndrome
Implantable haemodynamic monitors
Table 1. Implantable haemodynamic monitors.
| Device | Manufacturer | Access | Mechanism | Major inclusion criteria | Clinical evidence | Approval status |
|---|---|---|---|---|---|---|
| CardioMEMS | Abbott, USA | Venous, no septal puncture | Pressure monitoring in pulmonary artery | NYHA III, 1 HF hospitalisation in preceding year, GDMT with BB for at least 3 months, ACEi/ARB for at least 1 month | Reduction in HF hospitalisations, shorter LOS, improved QOL by KCCQ | FDA approved,CE marking |
| Cordella | Endotronix, USA | Venous, no septal puncture | Pressure monitoring in pulmonary artery | NYHA III, optimal GDMT for 3 months, HF hospitalisation, need for IV diuretics in the preceding year or elevated pro-BNP | Improvement in NYHA Functional Class, improved QOL by KCCQ | Under clinical trial (ClinicalTrials.gov:NCT04089059, NCT03375710) |
| V-LAP | Vectorious Medical Technologies, Israel | Venous, with septal puncture | Pressure monitoring in left atrium | NYHA III-IVa HF for at least 6 months, on optimal GDMT for at least 3 months, at least 1 HF hospitalisation in the preceding year | No clinical evidence to date | Under clinical trial (ClinicalTrials.gov:NCT03775161) |
| ACEi: angiotensin-converting enzyme inhibitors; ARB: angiotensin receptor blockers; BB: beta blockers; BNP: B-type natriuretic peptide; CE: European Conformity; FDA: US Food and Drug Administration; GDMT: guideline-directed medical therapy; HF: heart failure; IV: intravenous; KCCQ: Kansas City Cardiomyopathy Questionnaire; LOS: length of stay; NYHA: New York Heart Association; QOL: quality of life | ||||||
RATIONALE
Elevated left atrial pressure (LAP) and the ensuing pulmonary and peripheral congestion is the main driver of unplanned hospitalisations and overall poor outcomes in HF patients12 and contributes to an increased burden on the healthcare system. HF decompensation leading to hospitalisation is multifactorial, typically detected late and often associated with therapy-resistant acute decompensated heart failure (ADHF). Direct haemodynamic monitoring of pulmonary artery (PA) or left atrial (LA) pressure offers a paradigm shift in the management of symptomatic HF patients. Detection of a rise in PA or LA pressures offers a window of opportunity that allows clinicians to act proactively prior to clinical decompensation. Several implantable devices for invasive haemodynamic monitoring are currently in various stages of development and are discussed in this section.
CARDIOMEMS
CardioMEMS (Abbott) (Figure 1A) is an implantable PA pressure monitor that allows for real-time pulmonary pressure monitoring in patients with HF13. The device consists of a central sensor with nitinol loops on either end which are expanded to hold the device in position. Pressure readings are acquired in the resting supine position. The system allows physicians to act upon the PA pressure trends using a prespecified algorithm to maintain stable pressure. The system provides daily feedback informing the physician on the immediate impact of therapeutic measures. CardioMEMS is implanted via venous access and is advanced over a guidewire into the pulmonary artery (typically the left descending pulmonary artery), where it is deployed. The implant procedure is straightforward14. In routine right heart catheterisation, the target delivery site is identified using a pulmonary angiogram of the left PA at its downward turn in 2 projections (anteroposterior and 30° right anterior oblique/left anterior oblique [RAO/LAO]) serving as a roadmap for delivery. The ideal sensor position is at the inferior and lateral branch of the left PA. The target delivery site for implant delivery is required to have >7 mm clearance in a 30 degree angulated segment. Using 12 Fr femoral access, the sensor delivery system is brought up to the identified target site. Once in position, the sensor is released from a delivery tool and nitinol wires stabilise the implant in the target position. The sensor is non-obstructive to flow and typically endothelialises within 3 months; the sensor is calibrated during manufacturing and is equalised to the invasive PA pressure during the procedure without requiring further adjustments.
Indications for implantation include HF with preserved ejection fraction (HfpEF) or HfrEF with New York Heart Association (NYHA) Functional Class III symptoms, with at least one HF hospitalisation in the preceding 12 months and use of optimal guideline-directed medical therapy (GDMT) for at least 3 months. The landmark CHAMPION trial demonstrated that CardioMEMS-guided medical management in 550 patients with HF led to a 37% risk reduction in HF hospitalisation, shorter hospital length-of-stay and improved QOL (by the Kansas City Cardiomyopathy Questionnaire [KCCQ]), with a 1.4% risk of device-related complications13. In a subsequent postapproval study of 1,200 patients, there was a 57% risk reduction in HF hospitalisations at one year15. Furthermore, results from the recently published GUIDE-HF trial (prespecified COVID-19 adjusted analysis) are consistent with the findings of the CHAMPION trial and provide evidence that LAP monitoring with CardioMEMS may reduce hospitalisation in patients with NYHA Class II symptomatology and in those without an HF hospitalisation in the previous 12 months but with a persistently elevated outpatient B-type natriuretic peptide (BNP) or N-terminal-pro-BNP (NT-proBNP) level16.
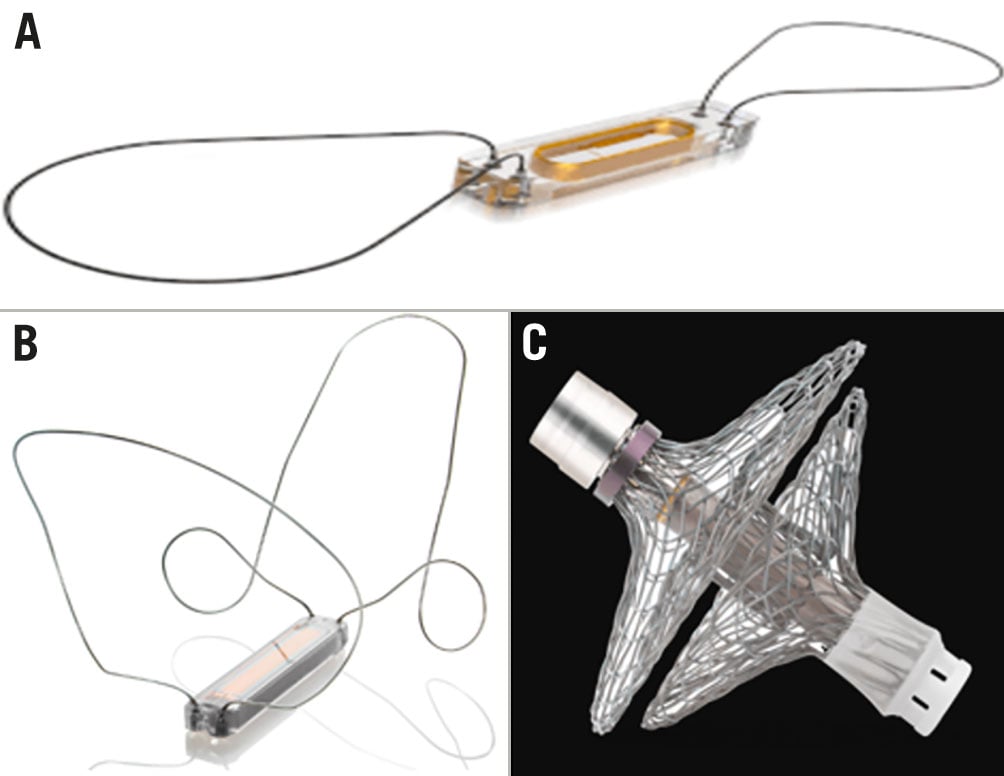
Figure 1. Implantable haemodynamic monitors. A) CardioMEMS (adapted from Abbott). B) Cordella (adapted from Endotronix). C) V-LAP (adapted from Vectorious Medical Technologies)
CORDELLA
The Cordella Pulmonary Artery Pressure Sensor System (Endotronix) (Figure 1B) is another implantable PA pressure monitor. The system consists of the commercially available Cordella Heart Failure System (CHFS) and the investigational Cordella PA Sensor System, aiming to provide comprehensive monitoring of PA pressure, body weight, blood pressure and oxygen saturation. The Cordella PA Sensor System comprises the PA sensor, the Cordella delivery system and a handheld reader. The PA sensor is a permanent implant flanked by two self-aligning nitinol anchors. The device is implanted via femoral venous access, the delivery system is guided to the right PA, and the device is deployed at the inferior-posterior inflection of the right PA with a typical vessel diameter of 12-26 mm. The handheld reader weighs 600 g and allows for pressure acquisition in both seated and supine positions using the handheld patient reader for 18 seconds to obtain PA pressure waveforms. These data, along with the vital signs, are securely transmitted from the tablet via the Cordella data analysis platform to the web-based application allowing remote viewing of daily submissions by a clinician or HF management team. The display allows simultaneous observation of supine and seated pressures, along with mean and systolic/diastolic PA pressure trends. This conceptually provides the added value of recording the haemodynamic changes not only in resting conditions but also during or after routine physical activity, such as exercise, or assessment of the haemodynamics during the standardised 6-min walk test (6-MWT). By incorporating non-invasive monitoring of vital parameters, Cordella offers comprehensive digital management and facilitates patient self-education. Eligibility criteria utilised in the clinical trial programme include HfpEF or HfrEF with NYHA Class III symptoms, at least 1 HF-related hospitalisation in the preceding year, and at least 3 months of optimal GDMT. In an early feasibility study of 15 patients, medical therapy guided by the Cordella system demonstrated improvements in NYHA Functional Class and QOL (by the KCCQ), with no significant device-related complications17. The recently published18 CE-Mark SIRONA 2 trial validated the safety and accuracy of invasive PA pressure monitoring, demonstrating sustained equivalence of direct- and sensor-monitored PA pressure. The Cordella system is currently undergoing study in the pivotal PROACTIVE-HF trial in the US and the EU (ClinicalTrials.gov: NCT04089059).
IMPLANTABLE DIRECT LEFT ATRIAL PRESSURE MONITOR
V-LAP
V-LAP (Vectorious Medical Technologies) (Figure 1C) is a direct, batteryless, wireless LA pressure monitoring system. The system consists of a 3x16 mm sensor with a surrounding nitinol mesh anchor, which is implanted in the intra-atrial septum via a transeptal puncture through femoral venous access. The V-LAP system includes simple external electronics that allow the patient to monitor the pressures and transmit them to the cloud-based system. In addition, an app-based module gives the patient access to their haemodynamic data, enabling physician-directed patient self-management. A built-in algorithm enables the patient to adjust his diuretics once the pressure is out of the optimal range of the predefined therapeutic plan, much like a diabetic patient adjusts insulin-dosing in response to glucometer readings and physician-prescribed guidelines. Haemodynamic monitoring can also facilitate detection of cardiac arrhythmias. As the device is placed on the interatrial septum, it remains unclear if its presence might undermine future interventions requiring transseptal puncture, but certainly a device that monitors direct right atrial (RA) and LA pressures would be very desirable. Eligibility criteria for implantation used in the ongoing clinical trials includes HF for at least 6 months, with evidence of HF decompensation in the preceding year, and use of optimal GDMT. The system is currently being assessed in the VECTOR-HF first-in-human trial (ClinicalTrials.gov: NCT03775161) and the VECTOR II clinical trial.
Expert commentary:
Direct invasive haemodynamic monitoring offers a new paradigm in HF treatment aimed at maintaining ambulatory clinical stability by managing changes in PA or LA pressure. This provides a window for proactive intervention based on altered haemodynamic trends to stabilise filling pressures and prevent subsequent HF deterioration. Initial proof of concept, provided by the CardioMEMS programme, offered the first evidence of the benefit of such a management strategy on clinical outcomes and healthcare resources. Other devices under study offer additional value by providing granularity in haemodynamic changes during daily activities. Currently, devices for direct PA pressure as well as LA pressure monitoring are being tested, and clinical research will validate their individual role in HF management. Implantation procedures are straightforward and adoptable by certified and trained interventional cardiologists and interventional HF specialists. Haemodynamic invasive monitoring offers an attractive link to complementary utilisation of other wearables, and non-invasive monitoring enables more comprehensive home-based monitoring. The biggest challenge to these technologies will be ensuring reimbursement for outpatient monitoring to financially support the effort required to monitor and manage large groups of patients. The other challenge is management of the large quantity of gathered data and assurance of patient motivation to provide regular data collection to the management team. The use of artificial intelligence and automated algorithms to evaluate haemodynamics and create automated alerts will be essential to maximising the benefits of monitoring and preventing patient and physician fatigue with regard to these devices. Remote monitoring systems provide the opportunity for patient self-monitoring and self-management that maximise the benefits of proactive management aimed at maintenance of volume status tailored to the patient’s daily activities and fitness. This is now proactively pursued in the Vectorious clinical trial programme. One may envision that the implementation of patient-tailored self-management becomes a tool for a more efficient utilisation of remote monitoring technology by ensuring euvolemia by ad hoc titration of HF medications, thereby preventing HF hospitalisations and improving quality of life. Analogous to the success of home-based glucose monitoring in diabetes, it may evolve to a physician-directed but patient self-managed care, thereby reducing the load and pressure on human resources of dedicated HF clinics. It should be also noted that haemodynamic assessment of PA or LA pressure is only one component of HF management and should be considered within the entire spectrum of HF management addressing multiorgan interaction, particularly with kidneys or systemic neurohumoral activation. Emerging technologies addressing euvolemic status may utilise alternative approaches such as the monitoring of vena cava dilatation with or without direct pressure assessment, bi-atrial pressure monitoring, or algorithms to detect arrhythmias, as well as including other monitors such as electrolytes and pH.
Left atrial decompression
Table 2. Left atrial decompression devices.
| Device | Manufacturer | Access | Mechanism | Inclusion criteria | Clinical evidence | Shunt diameter | Approval status |
|---|---|---|---|---|---|---|---|
| Corvia IASD | Corvia Medical, USA | Femoral vein, with septal puncture | Left-to-right interatrial shunting, permanent implant | Symptomatic HF with EF ≥40%, NYHA II-IVa, at least 1 HF hospitalisation in the preceding year or use of IV diuretics, or elevated BNP, optimal GDMT for ≥1 month, PCWP ≥25 mmHg during exercise, PCWP ≥5 mmHg compared to RAP | No difference in cardiovascular death, HF hospitalisations, KCCQ score. Improvement in NYHA Functional Class | 8 mm | FDA Breakthrough Device designation, CE marking |
| V-Wave | V-Wave, Israel | Femoral vein, with septal puncture | Left-to-right interatrial shunting, permanent implant | HF with NYHA II-IVa, at least 1 HF hospitalisation in the preceding year or elevated BNP, on optimal GDMT | Improvement in NYHA Functional Class and KCCQ scores | 5 mm | FDA Breakthrough Device designation, CE marking |
| TASS | Edwards Lifesciences, USA | Right internal jugular vein, no septal puncture | Left-to-right interatrial shunting, permanent implant | HF with NYHA II-IVa, at least 1 HF hospitalisation in the preceding year or use of IV diuretics, optimal GDMT for at least 3 months, PCWP >15 mmHg at rest or >25 mmHg during exercise, LAP>RAP by 5 mmHg at rest or >10 mmHg during exercise | First-in-human trial: improvement in NYHA Functional Class, HF hospitalisations, PCWP, improvement in 6-MWD and KCCQ scores | 7 mm | No FDA approval or CE marking |
| AFR | Occlutech, Sweden | Femoral vein, with septal puncture | Left-to-right interatrial shunting, permanent implant | NYHA II-IVa HF with EF ≥15%, optimal GDMT for >6 months, HF hospitalisation in the preceding year, PCWP >15 mmHg at rest or >25 mmHg with hand grip | First-in-human trial:improvement in NYHA Functional Class, 6-MWD and KCCQ scores | 6, 8 or 10 mm | FDA Breakthrough Device designation, CE marking |
| AFR: atrial flow regulator; BNP: B-type natriuretic peptide; CE: European Conformity; EF: ejection fraction; FDA: US Food and Drug Administration; GDMT: guideline-directed medical therapy; HF: heart failure; IASD: interatrial shunt device; IV: intravenous; KCCQ: Kansas City Cardiomyopathy Questionnaire; LVEF: left ventricular EF; NYHA: New York Heart Association; PCWP: pulmonary capillary wedge pressure; RAP: right atrial pressure; TASS: transcatheter atrial shunt system; 6-MWD: 6-minute walk distance | |||||||
Rationale
In healthy individuals, there is an increase in stroke volume (SV), heart rate (HR) and cardiac output in response to exercise – accommodated by a compliant ventricle. Patients living with HF typically have impaired ventricular compliance with or without reduced ventricular contractility, leading to elevated LV end-diastolic pressure (LVEDP)12. This elevated LVEDP is translated to the left atrium (LA), causing elevated LA pressures and pulmonary vascular congestion – resulting in the classic HF symptoms of dyspnoea and orthopnoea12. Elevated LA pressure or its surrogate, i.e., pulmonary capillary wedge pressure (PCWP), has been shown to drive mortality in patients with HF12 and, thus, has emerged as a possible interventional target to alleviate the cascade of pathologically responsive pressure elevation in the cardiopulmonary circuit.
A possible solution to reduce LA pressure is controlled left-to-right interatrial shunting. In simulated models19 and in real-world data from patients with atrial septal defects (ASD)20, left-to-right interatrial shunting significantly reduces LA pressure in response to exercise. These findings provide the rationale for a transcatheter interventional approach to create an artificial interatrial shunt to blunt the rise in LA pressure and thereby improve exercise tolerance, with the potential of improving overall clinical outcomes. There are several interatrial shunt devices that can be divided into permanent implants or “leave nothing behind” procedures.
Corvia InterAtrial Shunt Device
The Corvia InterAtrial Shunt Device (Corvia Medical) (Figure 2A) was designed to offload the LA in patients with HFpEF with an LV ejection fraction (LVEF) ≥40% who experienced HF hospitalisation and remained symptomatic despite optimal guideline-directed therapy. The pathophysiological postulate guiding the clinical development of the Corvia device is the reduction of exercise-induced rise in LA pressure. This is reflected by key eligibility criteria requiring an elevated exercise-induced PCWP ≥25 mmHg, absence of severe pulmonary hypertension, preserved right ventricular (RV) function, and a PCWP-RA pressure gradient ≥5 mmHg on haemodynamics obtained during a standardised supine exercise protocol21. The device consists of a 19 mm outer frame and creates a permanent 8 mm interatrial shunt. The device is implanted using femoral venous access and deployed after an atrial septal puncture.
Initial findings from the single-arm, CE-marked, randomised, mechanistic, sham-controlled REDUCE-LAP-HF trial demonstrated the safety and efficacy of the InterAtrial Shunt Device21 with a significant decrease in PCWP during exercise in the treatment arm. Early signals of clinical benefit demonstrated a lower incidence of HF admissions requiring intravenous diuretics and reduced mortality as compared to predicted mortality2122. However, the recently published sham-controlled REDUCE LAP-HF II trial23 yielded neutral results, with no difference in cardiovascular death (in fact an increase in major adverse cardiovascular events compared to the control group), HF hospitalisations, or KCCQ scores. Notably, in the overall patient population, a signal of early hazard for heart failure hospitalisation has been noted in patients with the shunt implant up to 12-month follow-up. In the post hoc analyses, a signal of benefit was noted in patients with low pulmonary vascular resistance (PVR) at peak exercise (PVR <1.74 Wood Units), accompanied by an improvement in NYHA Functional Class and quality of life. Sensitivity analyses also indicated signals of benefit in females and in patients without extreme right atrial dilation or pulmonary hypertension at rest. The RESPONDER-HF Trial (ClinicalTrials.gov: NCT05425459), a confirmatory study evaluating the effects of this shunt in patients like those identified in the post hoc analysis, is soon to be launched. Discussion on the significance of these results can be found in the “expert commentary” section below.

Figure 2. Interatrial shunt devices. A) InterAtrial Shunt Device (adapted from Corvia Medical). B) V-Wave Shunt System (adapted from V-Wave). C) Atrial Flow Regulator (adapted from Occlutech). D) Transcatheter Atrial Shunt System (adapted from Edwards Lifesciences)
V-Wave Device
The V-Wave Ventura Interatrial Shunt System (V-Wave) (Figure 2B) is another prosthetic interatrial shunt device designed to lower LA pressure. The device is being clinically tested in a broader HF population including both HFrEF and HFpEF patients with NYHA Class II-IVa symptoms with prior HF hospitalisation or elevated BNP/pro-BNP (both NYHA Class II) and use of optimal GDMT. The device has an hourglass shape, with a narrow neck (5 mm shunt size) and broad inlets to both atria. It is encapsulated with polytetrafluoroethylene to limit tissue growth. The device is implanted via femoral venous access, and after transseptal puncture, the device is positioned under transoesophageal echocardiography (TOE) guidance.
The first-in-human experience24 in 38 patients evaluated the initial design with a tri-leaflet one-way valve; this appeared to be associated with high rates of device occlusion (14%) and device stenosis (36%). Nevertheless, the study demonstrated improvements in cardiac haemodynamics associated with improved symptoms, exercise tolerance and major adverse cardiac events. The original device has been modified and the one-way valve has been eliminated from its second iteration. The second-generation device is currently being evaluated in a randomised clinical trial to assess its efficacy in patients with HFrEF and HFpEF (ClinicalTrials.gov: NCT03499236). In the large, 97-patient roll-in cohort of this trial, the device has demonstrated excellent implant success and safety, 100% shunt patency through 12 months, and preliminary signals for efficacy including large, sustained improvements in KCCQ score (Bayes-Genis, A. Clinical Trials Update Presentation [Conference session]. European Society of Cardiology 2021 Digital Congress, Virtual Presentation. July 2021) and improvements in left and right ventricular structure and function (Nunez Villota J, et al. Improved left and right ventricular structure and function with the V-Wave Ventura Shunt Device in patients with HFrEF and HFpEF: 12-month echocardiographic results from the RELIEVE-HF roll-in cohort. ESC Heart Failure 2022, Madrid, Spain. Late Breaking Clinical Science presentation, May 21, 2022).
Atrial Flow Regulator
The Atrial Flow Regulator (Occlutech) (Figure 2C) is another device designed to reduce LA pressure through interatrial shunting. The device is composed of two flat nitinol disks and is manufactured in three different shunt sizes (6, 8 or 10 mm). Access is obtained via the right femoral vein, a transseptal puncture is performed, and the device is deployed following balloon atrial septostomy – given that this device has less radial force than the aforementioned devices25. The device design allows highly controlled deployment with the possibility of retrieval after the final step of its positioning on the right atrial side. Main eligibility criteria used in the early clinical testing include symptomatic HF with NYHA Class III-IVa, HFpEF or HFrEF (EF ≥15%), with at least one HF hospitalisation in the preceding year, use of optimal GDMT for at least 6 months, and PCWP ≥15 mmHg at rest or ≥25 mmHg during exercise. In the initial clinical testing, the degree of filling pressures at rest or during exercise served as guidance for the choice of shunt size, with a 10 mm size implanted in patients with induced PCWP increase as compared to an 8 mm shunt size used in patients with elevated resting filling pressures. The device can be tailored to the septal thickness, offering either a 5 or 10 mm height. In an early feasibility study of 36 patients with both HFpEF and HFrEF, there was significant improvement in NYHA Functional Class, QOL (by the KCCQ), 6-min walk distance (6-MWD), HF admission and PCWP at 3-month follow-up25. These findings were further corroborated at one-year follow-up in a cohort of 53 non-randomised patients, supporting sustained shunt patency and its clinical efficacy with improved symptoms and exercise tolerance26. No safety concerns were noted across the heart failure phenotypes treated with either 8 or 10 mm shunts. A pivotal clinical trial is scheduled to begin during the second half of 2022 (ClinicalTrial.gov: NCT05136820).
Transcatheter Atrial Shunt System (TASS)
The Transcatheter Atrial Shunt System (TASS; Edwards Lifesciences) (Figure 2D) is an atrial shunt device with left-to-right shunting via the coronary sinus (CS). The device has 4 arms (2 in the LA and 2 in the CS) and a shunt diameter of 7 mm. Access is obtained through the right internal jugular vein, and a wire is advanced into the coronary sinus from where the LA is accessed. Important eligibility criteria include symptomatic HF (HFpEF or HFrEF) with NYHA Class II-IVa (with HF admission or use of high-dose diuretics in the preceding 12 months), PCWP >15 mmHg at rest or >25 mmHg during exercise, along with LA pressure exceeding RA pressure (5 mmHg at rest or 10 mmHg during exercise).
The first-in-human trial with 11 patients using the TASS (3 were not implanted due to anatomical factors) showed significant improvement in NYHA Functional Class, reduction in HF admissions, along with improved PCWP, QOL and 6-MWD at 6-month follow-up (Simard T, et al. TCT-87 Levoatrial to coronary sinus shunting as a novel strategy for symptomatic heart failure: First-in-Human experience. J Am Coll Cardiol. 2019;74:B87)
The device is currently being evaluated in an early feasibility study (ClinicalTrials.gov: NCT03523416).
NoYA: Adjustable interatrial shunt device
The NoYA system (NOYA MedTech) belongs to a class of devices designed to “leave nothing behind”. It is a radiofrequency ablation catheter designed as a self-expandable nitinol stent with an electric pole on the waist. The electric pole ablates the atrial septal tissue and adjusting strings are used to adjust the diameter within a 4-12 mm range during the procedure. The shunt device is removed after the procedure and effectively creates an ASD. In the experimental proof of concept, the intervention appeared safe with sustained patency and no thromboembolic or other complications. The first-in-human experience, limited to 10 patients, demonstrated early feasibility and favourable changes in surrogate efficacy parameters; however, shunt narrowing over time was reported in the majority of patients27. An additional clinical programme is under development to evaluate the efficacy and safety of this approach in a larger patient population.
Alleviant
Alleviant (Alleviant Medical) is another “leave nothing behind” interatrial shunt system. Access is obtained via the femoral vein, and a catheter with a blade at the distal tip is advanced to the interatrial septum. Septal tissue is excised and removed using echocardiographic and fluoroscopic guidance, creating a shunt. Eligibility criteria include NYHA Class II-IVa symptoms, LVEF >40%, and hospitalisation or use of intravenous diuretics for HF within 12 months prior to the procedure. A first-in-human study of 10 patients demonstrated improvements in PCWP, 6-MWT, pro-BNP, KCCQ and NYHA Functional Class (Barker CM et al. Novel no-implant interatrial shunt for heart failure: First in human clinical experience. J Am Coll Cardiol. 2021;77:1094). Alleviant had a 100% success rate and no procedure-related complications at 30 days. A clinical trial is currently underway to assess the safety and efficacy of the device (ClinicalTrials.gov: NCT04583527), and a pivotal trial is in the planning stage.
InterShunt
InterShunt (InterShunt Technologies) is a percutaneous atrial shunt catheter system that also uses the “leave nothing behind” approach. Access is obtained via the femoral vein, and the system is advanced to the atrial septum, where septal tissue is excised and removed to create a shunt. The device is in its early design stages, and clinical data are currently pending.
Expert commentary:
Transcatheter LA decompression has emerged as a promising intervention to address the key mechanistic cornerstone leading to the cascade of heart failure-related congestion. The rationale for LA decompression is based on data supporting the prognostic relevance of increased LA pressure and improved outcomes once LA pressure is successfully reduced by medical intervention28. It should also be noted that the concept of LA decompression is also soundly supported by the pathophysiology of volume overload. Here, LA decompression can complement diuretic regimens, especially in cases of fast, vascular-dependent volume overload. Initial criteria for patient selection or choice of shunt size are largely based on the computer modelling obtained in HFpEF patients during standardised invasive exercise haemodynamics19. The presence of left-to-right pressure gradient is required to elicit haemodynamic improvement by reducing the left-sided filling pressures. As this leads to increased left-to-right blood flow, patients with substantially reduced right heart function with elevated right atrial pressures or severe pulmonary hypertension should be avoided. However, based on these basic haemodynamic selection criteria and modelling-predicted haemodynamic benefit, the pivotal REDUCE LAP-HF trial failed to reach its primary endpoint using the 8 mm shunt size. Nevertheless, the post hoc analyses pointed at clinical benefit in subsets of HFpEF patients, in particular females and, as hypothesised by prespecified sensitivity analyses, in the absence of severe pulmonary hypertension or right heart dilation. Post hoc analysis revealed a provocative insight, suggesting an absence of benefit in patients with latent pulmonary vascular disease. This new index has been defined as an absence of increased pulmonary vascular resistance during standardised supine exercise. An additional, provocative finding has been the notion of clinical hazard in patients with a pacemaker. Unfortunately, the published data do not provide a deeper understanding about the type or intensity of pacing, whether any type of right-sided pacing in the HFpEF subset is a pre-existing risk for shunting, and whether this could be identified by any functional or structural abnormalities in the screening phase.
Undoubtedly, the findings also point to the heterogeneity of the heart failure syndrome and the need to refine the identification of optimal responders which is being studied in several ongoing clinical trials. The fact that neutral signals came from the Corvia studies should not discourage clinicians nor industry from further exploring the benefits of LA decompression, which could potentially address an unmet clinical need across all heart failure phenotypes. Also, the concept of LA-to-RA decompression versus LA decompression into the coronary sinus may potentially have very different outcomes. It is of note that early signals of benefit have been demonstrated also in HFrEF patients whose selection did not require exercise haemodynamics and relied on documentation of a left-to-right gradient at rest. Here, experiences in the AFR-PRELIEVE and V-Wave roll-in phases support the potential clinical benefits. It is of note that the V-Wave shunt device has a smaller diameter (5 mm) raising the question about the optimal shunt size. Namely, the 5 mm shunt results in a >50% lower shunt flow at a 5 mm pressure difference between left- and right-sided filling pressure compared to an 8 mm shunt, such as that used by Corvia. This raises the hypothesis that a smaller shunt size, while related to a smaller shunt flow, might be haemodynamically effective enough to offload the left heart without compromising the pre-existing right heart structure and function. Experience from the non-randomised roll-in phase presented at recent scientific meetings corroborates this hypothesis by showing haemodynamic and clinical benefits and even improvements in right ventricular function. As the concept of LA decompression is based on computational modelling without the possibility of testing in the appropriate heart failure model, particularly in case of HFpEF, further research is needed to understand which clinical features may help identify optimal responders with or without exercise haemodynamics, the long-term impact of creating an iatrogenic atrial septal defect and the risk of paradoxical embolism. The field of LA decompression will remain undoubtedly dynamic. While exploring questions related to shunt size and the importance of exercise testing to identify optimal responders using resting haemodynamic or structural cardiac evaluation, we are likely to witness other studies exploring its role in specific disease settings such as symptomatic patients post-TAVI or after MitraClip. The latter is of particular importance as recent observational data point to worse outcomes in patients with haemodynamically significant left-to-right shunts after MitraClip intervention. Accordingly, the hypothesis that a well-controlled shunt as produced by the V-Wave shunt device with limited yet effective LA offloading may be clinically effective is currently being tested in the RELIEVE-HF randomised clinical trial.
Neuromodulation for management of heart failure
Table 3. Neuromodulation for HF.
| Target | Device | Manufacturer | Access/implant | Mechanism | Major inclusion criteria | Clinical evidence | Approval status |
|---|---|---|---|---|---|---|---|
| Baroreceptor activation | Barostim Neo | CVRx, USA | Pectoral pulse generator with electrode over carotid sinus | ↑parasympathetic activity, ↓sympathetic activity | Age ≥21, NYHA II-III, LVEF ≤35%, on GDMT for at least 1 month | Improvement in 6-MWD, pro-BNP levels, and QOL | FDA approved, CE marking |
| Thoracic baroreceptor | Harmony | Enopace, Israel | Endovascular implant into thoracic aorta | ↑parasympathetic activity,↓sympathetic activity | NYHA II-III on optimal GDMT | Pending | No FDA approval or CE marking |
| Vagal nerve stimulation | VITARIA | LivaNova, UK | Pectoral pulse generator with electrode over the vagus nerve | ↑parasympathetic activity, ↓sympathetic activity | NYHA II-III, LVEF ≤40%, LVEDD 5-8 cm | Improvement in NYHA Functional Class, LVEF, 6-MWD, QOL by MLHFQ | FDA Breakthrough Device designation, CE marking |
| Splanchnic nerve block | Satera Ablation System | Axon Therapies, USA | Femoral venous access with catheter ablation of splanchnic nerve in the 10th-11th thoracic vertebrae | Intravascular volume redistribution | NYHA II-IVa, LVEF >50%, PCWP ≥25 mmHg, on GDMT for >1 month | Pending | No FDA approval or CE marking |
| BNP: B-type natriuretic peptide; CE: European Conformity; FDA: US Food and Drug Administration; GDMT: guideline-directed medical therapy; HF: heart failure; LVEDD: left ventricular end-diastolic diameter; LVEF: left ventricular ejection fraction; MLHFQ: Minnesota Living with Heart Failure Questionnaire; NYHA: New York Heart Association; PCWP: pulmonary capillary wedge pressure; QOL: quality of life; 6-MWD: 6-minute walk distance | |||||||
BARORECEPTOR ACTIVATION THERAPY
Rationale
In patients living with HF, autonomic imbalance with increased sympathetic tone and decreased parasympathetic output is a maladaptive process, leading to increased myocardial workload accelerating further HF progression and decompensation29. Stimulation of baroreceptors can lead to decreased sympathetic activity and increase parasympathetic signals, resulting in a reduction in HR, decreased afterload, improved ventriculoarterial coupling and increased diuresis29. Likewise, direct vagus nerve stimulation (VNS) by increasing parasympathetic tone reduces HR, myocardial oxygen demand and afterload29. Several devices intervening at various anatomical levels of parasympathetic signalling and baroreceptors are in various stages of development.
Barostim Neo
Barostim Neo (CVRx), is an implantable pulse generator (placed in the pectoral region) connected to an electrode placed over the carotid sinus. The device stimulates the carotid baroreceptors to reduce sympathetic activity and increase parasympathetic tone, thus rebalancing the autonomic nervous system. The efficacy of the device has been assessed in the HOPE4HF and BeAT-HF trials. HOPE4HF30 was a randomised controlled trial with 146 patients who had NYHA Class III symptomatology and an LVEF ≤35%. At a mean follow-up of 6 months, the device demonstrated improvements in NYHA Functional Class, 6-MWD, pro-BNP levels, LVEF and QOL by the Minnesota Living with Heart Failure Questionnaire (MLHFQ)30. BeAT-HF31 was a subsequent pivotal study with 408 patients who had NYHA Class II-III symptoms and an LVEF ≤35%, which demonstrated improved QOL, 6-MWD and pro-BNP levels at 6-month follow-up. In 2019, the Barostim Neo received U.S. Food and Drug Administration (FDA) approval for the improvement of symptoms in patients who were not CRT candidates and had NYHA Class III symptoms, LVEF ≤35% and pro-BNP <1,600 pg/mL, based on the Phase 1 outcomes of the BeAT-HF trial. The ongoing postapproval Phase 2 of BeAT-HF is evaluating the effect of the Barostim Neo on morbidity and mortality, with results expected in 2023.
Harmony System
The Harmony System (Enopace) is an implantable system capable of delivering stimulation to the aortic wall consisting of an implantable unit, delivery catheter, patient wearable unit and remote programming unit. The implantable unit is composed of a nitinol stent-like device bearing 4 stimulation platinum/iridium electrodes, a receiving RF antenna coil (gold covered with ethylene/tetrafluoroethylene) and a titanium sealed electrical circuit unit and is implanted using endovascular techniques. The stimulation parameters can be remotely programmed using a dedicated wireless communication system and software. The mechanism of action relates to stimulation of aortic afferent fibres leading into the left vagal trunk, restoring autonomic balance with improved central blood pressure control, reduced arterial stiffness and improved cardiac performance. The individual patient responsiveness to reduce blood pressure in response to aortic stimulation is tested prior to implantation to optimise device positioning. The ongoing CE-Mark ENDO-HF study is targeting patients with HFpEF and HFrEF with a readout of safety and efficacy signals of cardiac structure and function as well as quality of life and exercise tolerance at 6 months.
VITARIA Vagus Nerve Stimulation Therapy System
The VITARIA System (LivaNova) is a VNS therapy device consisting of an impulse generator implanted in the right pectoralis muscle with a subcutaneous electrode that stimulates the vagus nerve. The system is designed to increase parasympathetic tone in patients with HF. In a pilot study of 60 patients (ANTHEM-HF32) with NYHA Class II-III symptoms, LVEF ≤40% and LV end-diastolic diameter 5-8 cm, treatment with the device showed significant improvements in NYHA Functional Class, LVEF, 6-MWD and QOL by MLHFQ. The device has since received FDA Breakthrough Device designation and is now undergoing two clinical trials (ANTHEM-HFrEF Pivotal Study, ClinicalTrials.gov: NCT03425422 and ANTHEM-HFpEF, ClinicalTrials.gov: NCT03163030).
SPLANCHNIC NERVE BLOCKADE
Rationale
The splanchnic vascular bed is an important physiological reservoir for intravascular volume distribution. It has classically been a target for the management of acute gastrointestinal bleeding in patients with cirrhosis and portal venous hypertension. Splanchnic nerve blockade (SNB) is a novel target for the management of patients with HF, given that SNB can lead to splanchnic vasodilation, reducing preload and afterload29.
Satera Ablation System
The Satera Ablation System (Axon Therapies) is an investigational device for SNB. In smaller studies of patients with HF, both percutaneous33 and surgical34 SNB led to significant improvements in cardiac filling pressures, cardiac output, and exercise capacity. The Satera Ablation System requires femoral venous access, after which the system is advanced to the splanchnic nerve in the 10th-11th thoracic vertebrae, and ablation is performed under fluoroscopic guidance. The system is currently undergoing a clinical trial with a goal to enrol 80 patients, age ≥40, HFpEF (EF >50%) on GDMT for 1 month, PCWP ≥25, and NYHA II-IVa symptoms (ClinicalTrials.gov: NCT04592445).
Expert commentary:
Interventions targeting neuromodulation are based on sound pathophysiological concepts with ample experimental evidence that stimulation of parasympathetic activity or activation of high-pressure baroreceptors – such as those in the carotid sinus – restore autonomic balance and improve cardiac haemodynamics. Neuromodulation strategies appear particularly fitted to addressing the fast or vascular pathway for increased preload where splanchnic blood volume redistribution leads to acute congestion and dyspnoea symptoms. This pathomechanism is not easily addressable by diuretics as such patients typically display euvolemic status during routine daily activities. Early evidence with neuromodulation strategies, either those restoring autonomic balance by vagal stimulation or by ablative intervention of the splanchnic bed, shows corroborative efficacy signals on long-term symptomatic endpoints with improved exercise tolerance and quality of life. It should be noted that the acute impact of these interventions lacks specific biomarker or procedural confirmation of long-term efficacy except for endovascular aortic thoracic stimulation. Here, periprocedural neurostimulation is an essential step in identifying the location of the implant, potentially helping to identify optimal long-term candidates for the therapy. Overall, further research is needed to identify optimal patient clinical profiles and stages of heart failure optimally benefiting from disruption of autonomic imbalance. In addition, refined tailoring of the neurostimulation algorithm while preventing tachyphylaxis should be explored to optimise the efficacy of neuromodulation.
Device-based therapy for cardiorenal syndrome
Rationale
In ADHF with cardiorenal syndrome (CRS), patients have reduced cardiac output, hypotension, and activation of the renin-angiotensin-aldosterone pathway, leading to vasoconstriction as a compensatory mechanism, resulting in decreased renal perfusion and diuretic resistance35. The insult from renal hypoperfusion is further exacerbated by an elevated central venous pressure (CVP) and consequent renal venous congestion36. Impaired renal arterial perfusion combined with renal venous congestion decreases the pressure gradient across the renal parenchyma leading to a decrease in glomerular filtration pressure36. This cycle of HF leading to renal dysfunction responds poorly to medical therapy and provides a significant challenge to the clinicians managing these patients. Described in this section are several catheter-based devices currently in development to increase renal perfusion (pushers) and decrease venous congestion (pullers).
Devices for renal artery perfusion (“pushers”)
Table 4. Devices for cardiorenal syndrome.
| Device | Manufacturer | Access/ mechanism |
Physiologic modification | Support | Major inclusion criteria | Clinical evidence | Regulatory approval |
|---|---|---|---|---|---|---|---|
| “Pushers”: increase renal artery perfusion | |||||||
| Aortix Percutaneous Mechanical Circulatory Support System | Procyrion, USA | Femoral artery/axial flow pump | ↑Renal perfusion, ↓cardiac afterload | Temporary, up to 5 L/min | Hospitalisation with ADHF, renal dysfunction, IV diuretics for >48 hours, PCWP ≥20 or CVP ≥12 | First-in-human study: 10X increase in urine output, no device related complications | FDA Breakthrough Device designation, no CE marking |
| Reitan Catheter Pump | Cardiobridge, Germany | Femoral artery/axial flow pump | ↑Renal perfusion, ↓cardiac afterload | Temporary | N/A | Efficacy study: improved urine output, cardiac index, serum creatine | No FDA approval or CE marking |
| Second Heart Assist Device | Second Heart Assist, USA | Femoral artery/axial flow pump | ↑Renal perfusion, ↓cardiac afterload | Temporary + long term, 4-6 L/min | N/A | No clinical evidence to date | No FDA approval or CE marking |
| “Pullers”: decrease renal venous congestion | |||||||
| Doraya Renal Flow Regular | Revamp, Israel | Femoral venous/partial occlusion of infra-renal vena cava | ↓Renal afterload, ↓cardiac preload | Temporary | ADHF with poor diuretic response | No clinical evidence to date | FDA Breakthrough Device designation, no CE marking |
| preCARDIA System | Abiomed, USA | Femoral venous/partial occlusion of superior vena cava at the right atrial junction | ↓Renal afterload, ↓cardiac preload | Temporary | NYHA III-VI, poor diuretic response | Proof-of-concept study with reduction in biventricular pressures | FDA Breakthrough Device designation, no CE marking |
| TRVD System | Magenta Medical, Israel | Femoral venous/axial flow pump | ↓Renal afterload, ↓cardiac preload | Temporary | ADHF with poor diuretic response, IVC >2 cm, elevated BNP, LVEF ≤40%, CVP >14 mmHg | First-in-human study with reduction in CVP | No FDA approval or CE marking |
| ADHF: acute decompensated heart failure; BNP: B-type natriuretic peptide; CE: European Conformity; CVP: central venous pressure; FDA: US Food and Drug Administration; IVC: inferior vena cava; LVEF: left ventricular ejection fraction; NYHA: New York Heart Association; TRVD: Transcatheter Renal Venous Decongestion | |||||||
Aortix
The Aortix Percutaneous Mechanical Circulatory Support System (Procyrion) (Figure 3A) is a device designed for temporary cardiac unloading and for renal perfusion augmentation. Aortix is a 6 mm axial flow pump, placed at a suprarenal position in the descending aorta via femoral arterial access, and provides flows of up to 5L. The device was tested in a first-in-human study enrolling 6 patients with HFrEF, who were undergoing complex PCI and required intra-procedural mechanical support. In this study, device placement took 4-9 minutes, for a mean support time of 70 minutes, and resulted in a mean 10-fold increase in urine output37. There were no device-related complications, including major bleeding or haemolysis. The device is currently undergoing a multicentre, non-randomised feasibility study in patients with ADHF and evidence of worsening renal dysfunction (ClinicalTrials.gov: NCT04145635). Important eligibility criteria include active hospitalisation for ADHF (HFpEF or HfrEF), with evidence of renal dysfunction (increase in Cr by ≥0.3 mg/dL) despite 48 hours of intravenous diuretics, venous congestion (PCWP ≥20 or CVP ≥12), along with exam findings suggestive of ADHF.
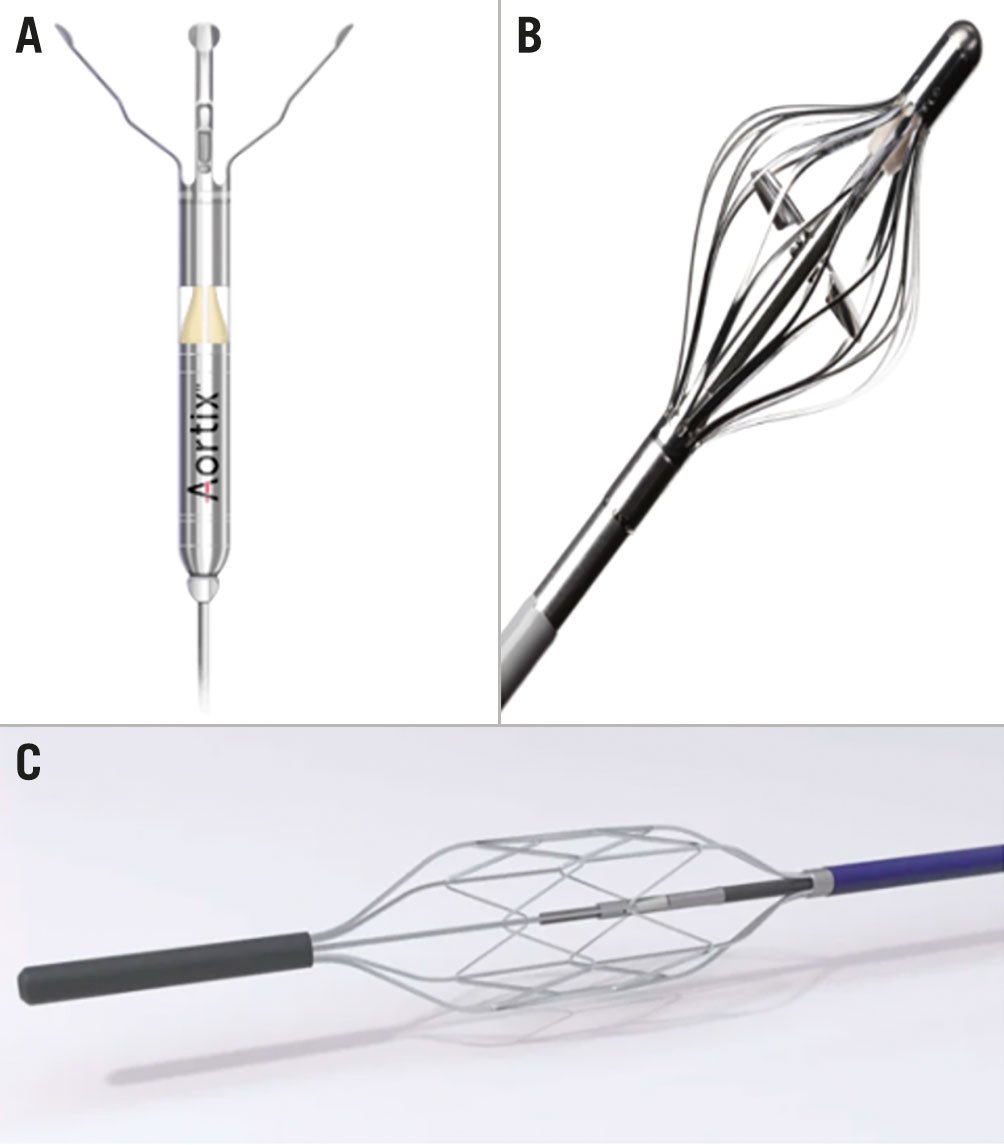
Figure 3. Devices for renal artery perfusion. A) Aortix Percutaneous Mechanical Circulatory Support System (adapted from Procyrion). B) Reitan Catheter Pump (adapted from Cardiobridge). C) Second Heart Assist Device (adapted from Second Heart Assist)
Reitan Catheter Pump
The Reitan Catheter Pump (Cardiobridge) (Figure 3B) is another device designed for the management of CRS in ADHF through afterload reduction and renal perfusion augmentation. It is a catheter-mounted pump with a collapsible 10 Fr head which is delivered to the descending aorta (5-10 cm distal to the left subclavian) via femoral arterial access. Once deployed, the pump is activated to achieve a radial-femoral gradient of 10 mmHg. The device was assessed in a study of 20 patients with an EF <30% who were admitted with ADHF and a cardiac index <2.1 L/min/m2 who required inotropic support38. After a mean support time of 18.3 hours, the mean cardiac index increased to 2.41 L/min/m2 (from 1.8 L/min/m2), along with an increase in urine output (from 71 mL/h to 227 mL/h) and a decrease in serum creatinine. There were no significant complications, including major bleeding or haemolysis38.
Second Heart Assist device
Another catheter-based solution to CRS is the Second Heart Assist device (Second Heart Assist) (Figure 3C). The pump has been developed in two designs: a catheter-based pump for temporary use (support for PCI, or treatment of CRS for 12-72 hours), and a wireless-powered implantable pump for management of chronic HF. The pump has an expandable 22 mm nitinol stent cage that expands to secure positioning, while allowing the device to be pulsatile compliant. Per the manufacturers, the device requires 35-50% lower pump speeds to generate outputs of 4-6 L to minimise haemolysis and mechanical wear. The device completed its preclinical studies in 2020, and there are currently no clinical data available.
Devices for renal afterload reduction (“pullers”)
Doraya Renal Flow Regulator
The Doraya Renal Flow Regulator (Revamp) is a temporary catheter-based device designed to reduce renal afterload and cardiac preload in patients with diuretic-resistant ADHF39. The device consists of a 25 mm nitinol frame that is introduced via the femoral vein into the infrarenal vena cava. The device partially restricts venous flow, creating an iliac venous pressure gradient of ~5 mmHg, thereby reducing renal afterload and cardiac preload with increased diuresis39. Per the manufacturers, the device is designed to be used for ≤12 hours and requires therapeutic anticoagulation. The FDA granted the device Breakthrough Device designation in 2020, and it is currently undergoing an early feasibility study (ClinicalTrials.gov: NCT03234647).
preCARDIA System
The preCARDIA System (Abiomed) is another device designed to reduce venous congestion. The device consists of a pump console and a catheter-guided balloon which is placed at the superior vena cava-RA junction and intermittently inflates to occlude flow to the RA, thereby reducing cardiac preload and renal afterload. In a proof-of-concept study of 8 patients with HFrEF, there was reduction in biventricular filling pressures, without any adverse events40. The device has been given Breakthrough Device designation by the FDA and is currently undergoing a feasibility study (ClinicalTrials.gov: NCT03836079) to evaluate the efficacy of the device in 30 patients with NYHA Class III-IV HF and evidence of inadequate diuresis.
TRVD system for renal venous decongestion
The TRVD system (Magenta Medical) is a catheter-based system that actively offloads the renal venous circulation by its positioning in a renal vein. A self-expanding, non-obstructive axial flow propeller-type pump is mounted on the distal tip of the TRVD catheter. The pump head includes a nitinol cage that protects the vein from the rotating propeller, which consists of a nitinol frame and silicone membrane. Target therapeutic pressures can be set separately for each renal vein. Renal vein pressure is actively reduced and controlled in an automatic closed-loop format to maintain the predefined target renal vein pressure within a narrow range. Preclinical and clinical data provided proof-of-concept evidence in improving urine output, increasing sodium excretion, and reducing central venous pressure. A first-in-human experience consistently showed immediate reduction in central venous pressure with improved renal blood flow41. Acute improvement in renal haemodynamics preceded a gradual decrease in right atrial pressures. Currently, a new-generation device is being developed to allow the placement of a single catheter into the inferior vena cava at the level of the renal veins that unloads the entire inferior vena cava segment, thereby simplifying the intervention without the need for selective renal vein cannulation and preprocedural imaging.
Other potential approaches for the management of CRS
In addition to the devices mentioned above, there are additional approaches under investigation for the management of CRS. One of these is cardiopulmonary nerve stimulation to enhance myocardial contractility and increase renal perfusion. Another promising approach is direct interstitial decongestion for concurrent interstitial and intravascular congestion. Conventional treatment of ADHF primarily targets intravascular fluid removal – even though most of the extracellular fluid is in the interstitial space and cleared via the lymphatic system – leading to hypotension from intravascular fluid depletion. The WhiteSwell System (WhiteSwell Medical) is a device designed to create a low-pressure zone in the thoracic duct to increase lymphatic flow, which has shown promise in an early feasibility study42. Direct interstitial decongestion may favourably affect the rise in renal interstitial pressure, allowing a potential avenue to manage patients with ADHF and CRS.
Expert commentary:
The granularity in understanding the pathophysiology of acute CRS led to novel device-based approaches to disrupt adverse pathophysiologic processes, in particular by targeting kidney haemodynamics and renal perfusion by altering renal venous afterload or arterial preload. Early studies provided clinical proof of concept in improving diuresis while preserving renal function. Further outcome studies are needed to facilitate their implementation in acute heart failure care.
Optimizer cardiac contractility modulation system
Cardiac contractility modulation (CCM) is another device-based therapy for chronic HF which is rapidly gaining traction and is now a part of the European Society of Cardiology Guidelines for management of HF. CCM therapy is designed to deliver high-voltage (~7.5 V), long-duration (~20 ms) electrical signals to the right ventricular septum during the absolute refractory period43. Therefore, it does not produce myocardial contraction, but rather results in changes in the molecular, cellular, and extracellular properties of the myocardium (believed to be through changes in calcium cycling) resulting in an increase in myocardial contractility and, over time, ventricular reverse remodelling.
The Optimizer smart system (Impulse Dynamics) consists of an implantable pulse generator, with standard pacing leads implanted in the RV septum. The device has been assessed in three randomised prospective trials, the most recent among them being the FIX-HF-5C trial44. The study consisted of 160 patients with an LVEF ≥25% and ≤45%, NYHA III-IV symptomatology on optimal GDMT and who demonstrated significant improvements in peak oxygen consumption, QOL by MLHFQ, NYHA Functional Class, and 6-MWD. There was also a significant reduction in the composite outcomes of HF hospitalisation and mortality.
Expert commentary:
CCM is an intriguing therapeutic modality with a unique mechanistic underpinning which is potentially synergistic and complementary to all other current approaches. The ultimate role and utilisation of CCM in the treatment of heart failure will likely be defined by two developments: first, the creation of a combined one-device platform for CCM and ICD for patients with a defibrillator indication; second, the ongoing AIM HIGHer trial (ClinicalTrials.gov: NCT05064709), a sham-controlled, randomised study with a primary composite endpoint of morbidity, mortality, and health status in patients with an LVEF of 40-60%.
Conclusions
There has been significant growth in the field of transcatheter valvular and non-valvular interventions for HF over the past decade. Recent advances in drug development addressing the well-characterised pathophysiological traits underlying heart failure has provided new pillars for current GDMT, positively impacting mortality and morbidity. Catheter-based interventions for HF are ambitious in offering potential solutions for the management of patients with HF who are unresponsive or intolerant to existing GDMT. The success of their clinical impact and adoption depends on several aspects. The basic prerequisite for successful device development is the identification of well-described pathophysiological targets in the setting of multiple pathophysiologically active pathways in heart failure. The target and proposed mechanism of action should be well documented and addressable against the background of patient-tailored GDMT or other established device interventions such as cardiac resynchronisation therapy. Heart failure patients are vulnerable and device development should deploy trial standards similar to those used in the development of transcatheter valvular interventions. These should also include local multidisciplinary heart failure teams capable of addressing the pros and cons of device intervention coupled with independent eligibility screening by an external committee. The choice of trial methodology and design is to be tailored to the target heart failure phenotype and underlying device mechanism of action. This requires the intensive interaction of the stakeholders as the bar for devices affecting hard clinical endpoints such as mortality may not be realistic for all interventions. Consistent with the recommendation of the regulatory authorities, trial endpoints should integrate patient-reported outcomes such as quality-of-life metrics or physical fitness that are validated and collected with the rigorous methodology of randomised and, when possible, sham-controlled clinical trials. The estimates of the improvement in these endpoints might be proposed as the main driver of the composite endpoints, without increased safety risk as evaluated by hard clinical endpoints. Clinical device development has more challenges than drug development as they need to demonstrate overall net clinical benefit while encompassing the procedural risk; moreover, long-term device performance and safety need to be proven. Currently, as devices need to demonstrate add-on benefit to patient-tailored GDMT, it must be seen whether, upon adoption, their clinical development may evolve on a par with medical therapy, in specific clinical settings or specific mechanisms of action. One example is the refined understanding of the volume overload in heart failure, where fast-response vascular overload is a currently elusive target for medical therapy. In this specific situation, LA decompression or neuromodulation may hypothetically provide a pathophysiologically sensible option for completing clinical testing. Clinical development should be stepwise, with primary proof of mechanism of action. Besides a randomised controlled design, clinical development should be granular and strive to identify the indication and timing of the given technology as compared to heart failure severity and stage, as well as comorbidities, in order to identify optimal responders and a window of opportunity for successful clinical adoption. In conclusion, we envisage that there will be a continued growth in transcatheter devices for heart failure to address complex pathophysiological mechanisms, either in conjunction with or to address pathways not fully addressed by medical therapy. These devices may be indicated in acute heart failure, in the prevention of acute decompensation of chronic heart failure, or in the progression of chronic heart failure.
Conflict of interest statement
A. Latib has served on advisory boards or as a consultant for Medtronic, Boston Scientific, Philips, Edwards Lifesciences, Abbott, and Ancora Heart. U.P. Jorde has served on advisory boards for Edwards Lifesciences and Abbott; has been a consultant for Edwards Lifesciences and Abbott and receives travel support from Ancora Heart. W.T. Abraham has received personal fees from Vectorious Medical Technologies and V-Wave. J. Bartunek serves on the Trial Steering Committee for Occlutech and as a member of the Clinical Events Committees and Data Safety Monitoring Boards of the Vectorious HF trials. He receives personal fees from Occlutech. M.H. Mustehsan has no conflicts of interest to declare.
