Abstract
Aims: Earlier generation self-expanding stents (SExS) showed high restenosis rates and long-term stent over-expansion. A novel SExS with reduced outward expansive force has been developed to overcome these limitations. This first-in-human study aimed to evaluate the safety and feasibility of the low pressure self-expanding nitinol-based vProtect™ luminal shield (LS) in the treatment of intermediate coronary lesions.
Methods and results: A total of 29 patients with clinical evidence of myocardial ischaemia and intermediate de novo coronary lesions were included. The LS was deployed after low-pressure balloon pre-dilatation. Acute procedural and device success was achieved in all patients. Angiographic follow-up at nine months showed an in-stent lumen loss of 0.50±0.30mm and a binary restenosis rate of 10.3%. There were no cases of late LS over-expansion or acute/late malapposition as evaluated by intravascular ultrasound ( IVUS). The cumulative major adverse cardiac events (MACE) rate at nine months was 10.3%, consisting of three target lesion revascularisations, with no cases of death, myocardial infarction or stent thrombosis.
Conclusions: Implantation of the LS in non-complex coronary lesions of intermediate severity was feasible, safe, and resulted in low rates of late loss and restenosis. IVUS analysis at nine months showed favourable mechanical properties of the LS without evidence of late device over-expansion.
Introduction
Balloon-expandable stent (BES) technologies are the standard of care for the interventional therapy of obstructive atherosclerotic coronary lesions1. Due to their mechanism of expansion, BES exert an unpredictable pattern of mechanical stress to the vessel wall leading to variable degrees of vascular injury and restenosis2. In addition, because BES rely on the plastic deformation of its structure via balloon dilatation, these devices are prone to acute under-expansion and late malapposition, especially in situations in which the vessel lumen cannot be accurately determined (e.g., ST-segment elevation myocardial infarction [STEMI]3).
Self-expanding stents (SExS) were developed to overcome these inherent mechanical limitations of BES, but demonstrated excessive radial force, resulting in late outward expansion and high restenosis rates4-6. New generations of self-expanding coronary stents displaying more stable biomechanics (lower chronic expansive forces) are under development7. These devices may improve clinical outcomes by providing suitable outward forces enabling proper vessel wall apposition and controlled luminal gain while reducing the amount of vascular injury, neo-intimal formation and positive vessel remodelling8. The resulting lower outward expansive force may also minimise the likelihood of inducing plaque rupture and distal embolisation at the time of implantation. In this first-in-human study, we sought to evaluate the feasibility and safety of implantation of a novel low pressure SExS vProtect™ luminal shield (LS) in patients with de novo intermediate coronary lesions.
Methods
Device description
The vProtect™ LS (Prescient Medical Inc., Doylestown, PA, USA) is a self-expanding nitinol-based stent, which has been designed to mechanically stabilise non-obstructive, coronary thin-cap fibro-atheromas. The LS exerts lower chronic outward forces than previous SExS while maintaining a stable radial force (crush-resistant), thus avoiding collapse following implantation. The device consists of the self-expanding LS and a rapid exchange delivery system. The LS has a strut thickness of ~57 microns and has a vessel surface area coverage from 13% to 15% in 2.5 to 3.0mm vessels7. The system is compatible with 0.014” guidewires and 6 Fr guiding catheters.
Study design
The current study was designed as a non-randomised, single-arm, single centre prospective trial to evaluate the feasibility and safety of implantation of the LS among patients with clinical evidence of myocardial ischaemia and intermediate de novo coronary lesions (so chosen to mimic the severity of borderline “vulnerable” lesions in which the device would subsequently be tested if the results of this experience were favourable). All patients had documented ischaemia either by clinical criteria, nuclear medical scans or stress echocardiography before they underwent cardiac catheterisation. Angiographic and intravascular ultrasound (IVUS) imaging were performed in all patients at baseline, post-procedure and at nine months follow-up. The study was approved by the Institutional Ethics Committee at Corbic Research Institute, Envigado, Colombia. All patients provided written informed consent for participation in the trial. The study case report for data was verified by independent study monitors (Clinlogix, North Wales, PA, USA). All potential adverse events were independently adjudicated by an independent clinical events committee and reported to the ethics committee.
Inclusion and exclusion criteria
Patients aged 18 years or older presenting with symptoms of coronary artery disease were eligible if they had a single, non-calcified target lesion by angiography of ≤15 mm in length with a diameter stenosis (DS) greater than 50% (by visual assessment) that was suitable for stent implantation in a vessel with a reference vessel diameter (RVD) ranging from 2.75 to 3.5mm in diameter. Only one LS was permitted to be implanted per lesion. Other lesions could be treated with other clinically approved devices.
The principal exclusion criteria were known allergy or sensitivity to nitinol or its components; known hypersensitivity or contraindications to anticoagulant or antiplatelet therapies; history of bleeding or known coagulopathy; major surgery within the past 30 days; lesions that were severely calcified based on IVUS imaging defined as a ring of calcium occupying more than 90 degrees of the lesion circumference; significant (>50%) left main coronary disease; previous stent placement or angioplasty in the target vessel; ST-segment elevation myocardial infarction; lesions involving a side branch of ≥2.0 mm in diameter or side branches <2.0mm with the presence of occlusive ostial disease or plaque shifting following balloon dilatation of the main vessel lesion; otherwise unsuitable coronary anatomy in the opinion of the investigator; females who were pregnant; participation in another investigational device or drug trial.
Procedural description
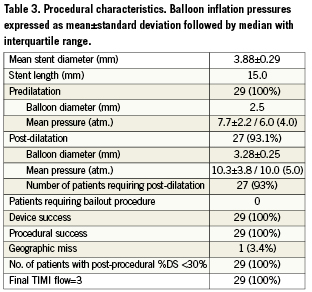
All patients received 600 mg of clopidogrel and 300 mg of aspirin (or 100 mg if they were already taking a daily chronic dose) at least two hours before the angioplasty procedure. Following the procedure clopidogrel was maintained at a dose of 75 mg for at least four weeks and aspirin was administered in a dose of 300 mg for at least one month. Aspirin was continued indefinitely at a dose of at least 100 mg daily. Intravenous unfractionated heparin was administered during the procedure to achieve and maintain an activated coagulation time (ACT) between 250 to 350 seconds. The use of glycoprotein IIb/IIIa antagonists was left to the discretion of the operator. Percutaneous coronary intervention was performed using standard techniques. The procedural methodology is shown in Figure 1, and a case example is shown in Figure 2. All target lesions were pre-dilated with a 2.5 mm balloon that was inflated in one atmosphere increments (“stepwise fashion”) until complete balloon dilatation was achieved. The target diameter of the LS was then selected based on quantitative coronary angiographic (QCA) measurements. The LS was available in diameters of 2.75 to 4.0 mm and in a single length of 15 mm. A LS was selected with diameter 0.5 mm greater than the RVD (e.g., 3.0 diameter for a ~2.5 mm RVD), and was deployed aiming to cover the borders of the pre-dilated lesion. Post-dilatation using a non-compliant balloon shorter than the total length of the LS was allowed when the residual on-line angiographic DS was ≥30%. The procedure was terminated when the angiographic DS was <30% and final TIMI 3 flow was achieved. Bailout stenting was performed if two consecutive balloon inflations failed to reduce the angiographic DS to <30% and/or reduced coronary flow or dissections were noted. A 12-lead electrocardiogram was obtained before the procedure, within 24 hours after and in the case of suspected acute ischaemia. Cardiac enzymes were monitored in all the patients following the procedure and before discharge.
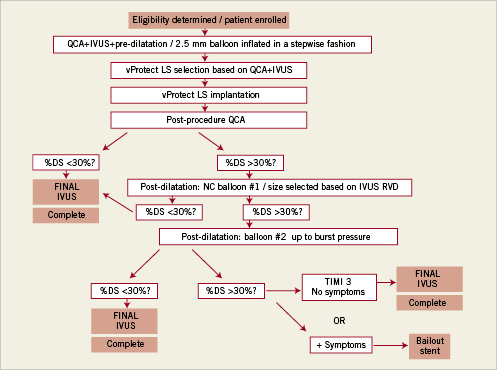
Figure 1. Flow chart of study design. NC: non-compliant
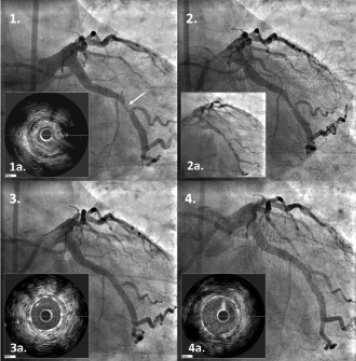
Figure 2. vProtect LS placement in the left circumflex (LCX) artery (LAO 0, CAU 20) of a patient undergoing PCI. 1. Baseline angiography presenting lesion in a mid-LCX (%DS=68%); 1a. IVUS cross-sectional view; 2. Effect after balloon pre-dilatation (2.5×9 mm; max. pressure: 12atm); 2a. vProtect luminal shield (4.0x15mm) positioning; 3. Final effect after vProtect placement and balloon post-dilatation (3.5×8 mm; max. pressure: 12atm); 3a. IVUS cross-sectional view of the final effect; 4. Angiographic results at nine-months follow-up; 4a. IVUS cross-sectional view at nine-month follow-up. Images courtesy of Corbic Research Institute, Envigado, Colombia
Study endpoints
Clinical follow-up was performed immediately after the procedure, prior to hospital discharge, at 30 days and at six, and nine months post-procedure. Angiographic and IVUS follow-up was performed in all patients at nine months following the procedure. The primary endpoints of the study were pre-specified as: 1) post-procedural angiographic DS ≤30%; 2) IVUS mean lumen area ≥4 mm2; and in-hospital and 30-day rates of major adverse cardiac events (MACE) defined as cardiac death, myocardial infarction (MI) or target lesion revascularisation (TLR). Secondary endpoints included six-month MACE and the nine-month rates of angiographic restenosis, TLR, target vessel revascularisation (TVR), target vessel failure (TVF, defined as death, MI or TVR) and MACE. TLR was defined as revascularisation (percutaneous intervention or bypass surgery) performed on the target lesion due to a stenosis including 5 mm margins (proximal and distal to the stent) at any time after the index procedure; TVR was defined as revascularisation performed on the target vessel at any time after the index procedure. A revascularisation procedure was adjudicated as “clinically indicated” if the DS of the treated lesion was ≥50% by QCA in the presence of ischaemic signs or symptoms, or if the DS was ≥70% irrespective of the presence or absence of ischaemic signs or symptoms. Device success was defined as attainment of ≤30% residual DS at the end of the procedure by using only the assigned device. Procedural success was defined as attainment of a ≤30% residual DS of the target lesion and freedom from in-hospital MACE.
Imaging analysis
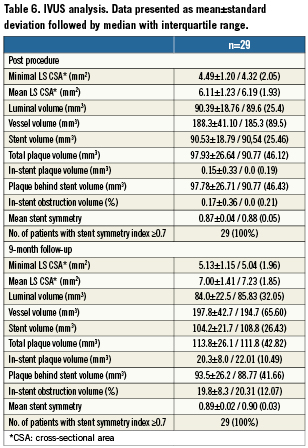
Angiographic and IVUS data were analysed by an independent core laboratory (Cardialysis, Rotterdam, The Netherlands). QCA analysis was performed with the CAAS II analysis system (Pie Medical BV, Maastricht, the Netherlands). All angiographic measurements were obtained within the stented segment (in-stent) and over the entire segment consisting of the stent and its 5 mm proximal and distal margins (in-segment). CURAD QCU Analysis Software (Curad B.V., Wijk bij Duurstede,The Netherlands) was used to analyse the IVUS images obtained immediately after stent implantation and at nine-month follow-up. Analysis was performed at the target segment (stent±5 mm) to measure and calculate the lumen, vessel, plaque and stent volumes. In addition, mean stent symmetry, in-stent obstruction volume(%) and number of patients with mean lumen area ≥4.0 mm2 were analysed.
Statistical analysis
All information was collected and processed in Excel database (Microsoft Corporation, Redmond, WA, USA). The following values were calculated: average (mean), standard deviation and percentage (whenever applicable). In addition, continuous angiographic and IVUS data were expressed as median with interquartile range.
Results
Study population
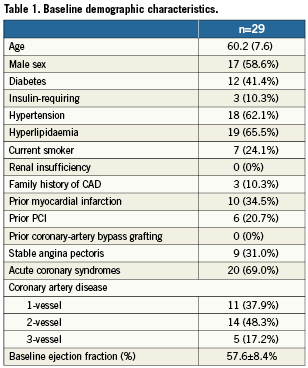
A total of 33 patients were screened and four were deregistered due to procedurally-related exclusion criteria. Baseline clinical variables are shown in Table 1. The mean age of the 29 patients was 60 years, and 59% were male. Demographic features were similar to most stent studies except for a higher proportion of diabetic patients (41%; 10.3% requiring insulin).
In-hospital outcomes
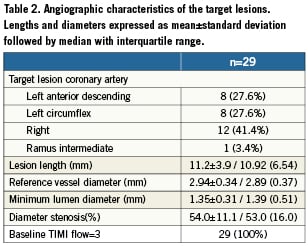
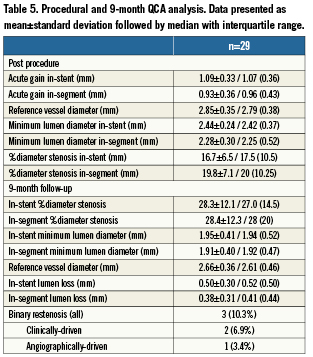
Baseline lesion characteristics and procedural data are presented in Tables 2 and 3. The mean baseline RVD was 2.94±0.34 mm, and the mean DS was 54.0%±11.1% (median %DS of 53.0 with interquartile range of 16.0), which decreased to 35.9%±8.2% immediately following LS implantation (p<0.0001) and then to 16.7%±6.5% after final balloon post-dilatation (p<0.0001). Post-dilatation was performed in 93% of patients (mean dilatation pressure: 10.3±3.8 atm) and achieved an average in-stent acute gain of 1.09mm (Table 3). A final DS<30% was achieved in all patients and device success was 100%. No patient required bailout stenting. There were no periprocedural complications and no occurrences of MACE, repeat revascularisation or stent thrombosis during the hospitalisation period.
IVUS analysis of luminal shield mechanics
The average minimal lumen area (MLA) before intervention in all enrolled patients was 2.75±0.74 mm2, and increased to 4.49±1.20 mm2 immediately after LS implantation and final balloon post-dilatation (a 63.3% increase from baseline, p<0.0001). All patients reached a post-procedural shield lumen area ≥4.0 mm2, and all had a shield symmetry index ≥0.7 (0.87±0.04). However, the lumen area tended to be slightly lower in the centre of the device where the plaque burden was the highest (Figure 3). Overall, the net acute volumetric luminal gain at the end of the procedure was 15% (Figure 4). There were no cases of acute stent malapposition either after the LS only or after post-dilatation.
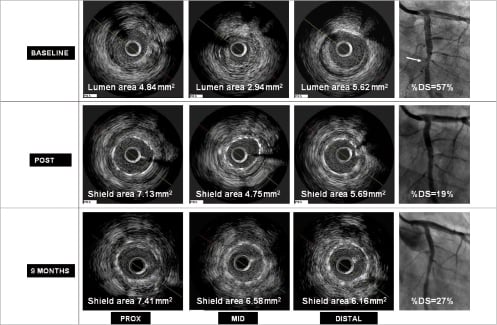
Figure 3. IVUS analysis of lumen and shield remodelling at proximal, mid and distal segments at different time points. Immediately following LS implantation the average lumen area was slightly lower in the centre of the device where the plaque burden was the highest. During the next 9 months the shield continued to enlarge resulting in a final lumen area comparable to what has been reported in BES studies with optimal device symmetry. The right column represents angiographic views of the mid LCX which correspond to the IVUS images on the left (RAO 30, CRA 0; the white arrow shows the lesion before the LS implantation). Images courtesy of Corbic Research Institute, Envigado, Colombia
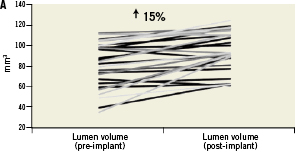
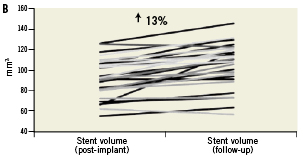
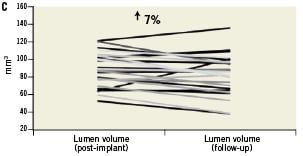
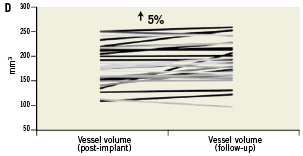
Figure 4. Mechanical behaviour of the vProtect™ LS and its influence on vessel and lumen remodelling based on IVUS calculations. A. Acute gain (difference between lumen volume pre-implant and lumen volume post-implant); B. Stent volume change (difference between stent volume post-procedure and stent volume at nine-month follow-up); C. Lumen volume change (difference between lumen volume post-procedure and lumen volume at nine-month follow-up); D. Vessel volume change (difference between vessel volume post-procedure and vessel volume at nine-month follow-up).
Long-term outcomes
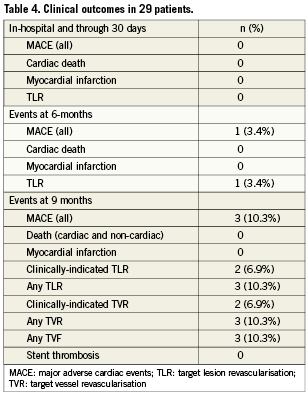
Clinical, angiographic and IVUS follow-up at nine months was completed in all enrolled patients. During nine-month follow-up no patient died, developed myocardial infarction or stent thrombosis (Table 4). In total, there were three cases of MACE, all TLR (10.3%). Two patients with TLR presented with symptoms of unstable angina, two and seven months following luminal shield implantation. In both of these patients angiographic in-stent restenosis was successfully treated with drug-eluting stents (DES). These two cases were adjudicated as a clinically-driven TLR. The third patient with TLR underwent successful PCI with DES implantation after the planned nine-month control angiogram revealed significant in-stent restenosis isolated to the edge of the LS. By QCA (Table 5), the mean in-stent late loss (LL) at nine-month follow-up was 0.50mm, the mean in-stent DS was 28.3%, and the total binary restenosis rate was 10.3%. There were no cases of late angiographic thrombi, aneurysm formation or LS fracture.
IVUS analysis of vascular remodelling
At nine months, IVUS volumetric analysis displayed an additional 13% increase in the total LS volume compared to the post-implantation values. The average minimal LS area at nine months was 5.13±1.15 mm2 (an additional 12.5% increase compared to the post-implantation time point; p=0.04). By volumetric analysis there was a non-significant 5% increase in total vessel volume over time (from mean 188.3 mm3 to 197.8 mm3, p=0.39) (Table 6 and Figure 4). The in-stent percent volume obstruction was 19.8% and the mean stent symmetry index was 0.89. There were no cases of late stent malapposition.
Discussion
Currently available balloon expandable stents rely on the plastic deformation of their metallic structure via mechanical expansion9,10. This mechanism of stent delivery elicits an unpredictable degree of mechanical injury to the vessel wall, resulting in an injury-dependent pattern of vascular healing and restenosis8. Stent malapposition frequently occurs immediately following BES deployment, especially when the luminal dimensions of the vessel cannot be accurately determined (e.g., STEMI)3. Late acquired stent malapposition can also occur during follow-up after DES implantation due to positive remodelling3,11,12, and has been associated with late stent thrombosis13. Due to their intrinsic mechanical properties and material composition, SExS technologies have the potential to overcome these limitations6. Specifically, SExS have favourable mechanical characteristics for treatment of coronary lesions in which either high-radial expansive forces are not required (i.e., non-calcified stenoses), stent-wall apposition is difficult to achieve and highly desirable (i.e., STEMI), or the potential for distal embolisation during PCI is likely (i.e., large necrotic cores)14-18. Early clinical experiences have also been reported with other SExS concepts among patients presenting with STEMI19 and ischaemic coronary artery disease20.
In the present study we evaluated the safety and efficacy of implantation a new SExS, the vProtect LS, for the therapy of intermediate lesions among patients with documented myocardial ischaemia scheduled for PCI. As has been previously published, the device utilised in this study is structurally and mechanically different than previous generations of SExS technologies21. The LS maintains lower and more stable chronic outward expanding forces over time than previous SExS, thus resulting in less vascular injury7,22. Specifically, the LS delivers 50% less chronic outward forces than the first generation SExS designs, while maintaining a similar radial resistance force at the same dilation diameters2. Pre-clinical studies demonstrated appropriate vascular healing with no evidence of chronic over-expansion at 90 days in normal porcine coronary arteries23.
Acute deployment of the LS resulted in 100% procedural success with no in-hospital or short-term clinical adverse events. By nine months, in-stent late loss was only 0.50 mm, and binary restenosis had occurred in three patients (10.3%), which is lower than described with first generation SExS platforms4,5. The three episodes of restenosis triggered three ischaemia-driven TLR procedures, two of which were clinically-driven. There were no occurrences of death, myocardial infarction or stent thrombosis during follow-up. Thus, MACE at nine months occurred in only three patients (10.3%).
Baseline, post-procedure and follow-up IVUS revealed important mechanistic insights to the LS performance and vascular responses. Following LS implantation, the device was always apposed to the vessel wall and conformed to different degrees of plaque burden, displaying lower luminal areas at places in which the plaque burden was the highest. In addition, the LS had a favourable post-procedure symmetry index, demonstrating homogeneous distribution of the radial force and no localised stent recoil. However, the ~17% residual in-stent DS with the LS is higher than that typically seen in BES trials, and by IVUS the final mean cross-sectional lumen area achieved (6.11±1.23 mm2; Table 6) was slightly lower than the average mean lumen areas reported with BES (range 6.5 to 9.3 mm3)24,25 and first generation SExS (7.7±2.1mm2)25.
During follow-up, gradual expansion of the LS (an additional ~13% minimal area gain over nine months) resulted in a final nine-months mean luminal area of 7.00±1.41 mm2 comparable to mean areas reported in prior BES studies (mean 7.55 mm; range: 6.5-9.4)24-26. Importantly, in contrast to earlier generation SExS, which were characterised by over-expansion and positive remodelling25, the late increase in LS area was not associated with a significant increase in total vessel dimensions. Instead, vascular remodelling over time with the LS was due to plaque remodelling over the length of the treated segment. Conversely, first generation SExS achieved stent areas comparable to BES almost immediately following stent implantation, after which over-expansion occurred (20% to 40% in stent areas) during the next six months4-6,25. Such chronic outward expansion with ongoing vascular injury was likely responsible for the higher rates of restenosis (17-24%) and late LL (0.82-0.98 mm) at six-month follow-up seen with these early SExS platforms4,5,25. However, although the encouraging late angiographic results with the LS may be due to the mechanical properties of the device itself, either the technique employed for plaque and device dilatation (“gentle balloon dilatation”) and/or lesion selection may have contributed to the favourable results observed. Of note, one of the three cases of restenosis with the LS was related to stent misplacement (“geographical miss”) resulting in edge restenosis rather than true in-stent neo-intimal hyperplasia.
The major limitations of the present study are the small sample size and the highly selected patient and lesion cohort that was enrolled. Specifically, the mean baseline DS of the studied lesions (54%) was less than usually seen in most stent studies (~64-68%), and calcified lesions were strictly excluded. A strength of the present study, however, is that clinical, angiographic and IVUS follow-up was completed in 100% of patients and at a longer follow-up time (nine months) compared to other BMS platforms4-6,27.
In summary, implantation of an innovative self-expanding nitinol-based stent (the vProtect LS) in non-calcified coronary lesions of intermediate severity is feasible, safe and resulted in a low rate of angiographic and clinical restenosis for a bare metal stent platform. The device maintained its mechanical integrity following implantation and resisted plaque compressive forces. During the nine months of follow-up the LS induced progressive but well-controlled plaque and vascular remodelling to a far less degree than previously reported with first generation SExS. Due to its intrinsic mechanical properties, the rates of angiographic late loss and binary restenosis may be lower than that seen with bare metal BES and earlier SExS, although comparative trials are required for confirmation. Given the mechanical properties of the LS, further studies are warranted to determine whether this device might be of particular benefit in specific subsets such as rupture-prone intermediate lesions with large necrotic core (“vulnerable plaques”), and in patients with acute coronary syndromes.
Conflict of interest statement
The authors have no conflict of interest to declare.
