
Abstract
Aims: This study sought to investigate outcomes of transcatheter aortic valve implantation (TAVI) with the SAPIEN 3 transcatheter heart valve (THV) in patients at intermediate risk for surgery. The 30-day results of the transfemoral cohort are reported.
Methods and results: The SAPIEN 3 European approval trial intermediate-risk cohort included a total of 101 patients with severe, symptomatic aortic stenosis, at intermediate risk for surgery suitable for TAVI via the transfemoral route (TF). Outcomes were adjudicated by a clinical events committee. Echocardiography, computed tomography and electrocardiography exams were analysed in core laboratories. The mean STS-PROM score and logistic EuroSCORE of the study population were 5.2±1.7 and 13.2±3.8, respectively. A completely percutaneous procedure was performed in 90.1% of patients and conscious sedation and/or local anaesthesia was utilised in 54.5%. Technical success was achieved in 98.0% of patients. At 30 days, mortality was 1.0%, with stroke in 3.0% and a new permanent pacemaker in 4.0% (4.3% of patients without pre-procedural permanent pacemaker). No patients had severe aortic regurgitation after the procedure, only one patient had moderate aortic regurgitation, and 70.8% of patients had no or trace aortic regurgitation.
Conclusions: TF-TAVI using the SAPIEN 3 THV in patients at intermediate risk for surgery is associated with a very low risk of death and complications, including new pacemakers and paravalvular leaks. Although compelling, these initial results are being confirmed in larger global studies before expanding the indications for TAVI in severe aortic stenosis. Clinical Trial Registration: https://clinicaltrials.gov/show/NCT01808287
Introduction
Transcatheter aortic valve implantation (TAVI) has gained widespread use in the management of patients with severe aortic stenosis (AS) and has rapidly become a routine clinical procedure in many centres. On the basis of the existing evidence1-3, guidelines recommend TAVI for the treatment of inoperable or high surgical risk patients4,5. However, the technology has rapidly evolved and the rate of periprocedural complications has dramatically decreased6, supporting progression of TAVI indications to include lower operative risk populations7,8. Prior studies have reported promising results in patients at intermediate surgical risk9-13. Nonetheless, these studies are retrospective, mostly using second-generation devices with both transfemoral (TF) and transapical (TA) access routes, and provided limited information as to the determinants of surgical risk8.
The last few years have witnessed exciting improvements in TAVI technology with the development of new-generation devices14. The SAPIEN 3 transcatheter heart valve (THV) is the next-generation device of the Edwards SAPIEN balloon-expandable valve family (Edwards Lifesciences, Irvine, CA, USA). Results of the initial experience with the SAPIEN 3 device in patients at high risk for surgery have already been reported15. In this current analysis, the 30-day outcomes of the SAPIEN 3 Transcatheter Heart Valve using the TF delivery route on patients at intermediate risk for surgery (Safety and Performance Study of the Edwards SAPIEN 3 Transcatheter Heart Valve/ The SAPIEN 3 Study [SAPIEN 3]) are reported.
Methods
POPULATION
Patients aged >75 years, with AS and symptoms (New York Heart Association [NYHA] Class ≥II) at intermediate risk for surgery were screened for inclusion in the study. Aortic valve stenosis was defined as an aortic valve area <1.0 cm2 and a mean gradient >40 mmHg. Intermediate risk for surgery was determined as a predicted mortality ≥4 and ≤8% by the Society of Thoracic Surgeons (STS)-PROM score or ≥10 and ≤15% by the logistic European System for Cardiac Operative Risk Evaluation (EuroSCORE) or on the basis of the assessment by the Heart Team. The delivery approach was determined by the Heart Team according to anatomical conditions and local practice. Only patients considered candidates for TAVI via the TF route are described in these study extension results.
The main exclusion criteria were the occurrence of a myocardial infarction within one month before the intervention, the presence of a bicuspid aortic valve, severe aortic regurgitation (AR), prosthetic valve in any position, a left ventricular ejection fraction <20%, the occurrence of a stroke or transient ischaemic attack within three months of the procedure, a creatinine level of >3.0 mg/dL or dialysis, estimated life expectancy <24 months and untreated pre-existing conduction disturbances (atrioventricular block [AVB] and bundle branch block) (Appendix). However, the last criterion was added after the trial was started, and a total of nine patients with either untreated left or right bundle branch block were included in the study, three patients who were included under the original protocol and six who were enrolled under the amended protocol and were considered protocol deviations.
STUDY DESIGN
The SAPIEN 3 Study design has been previously reported15. It is a non-randomised, prospective, multicentre study, including patients undergoing TAVI with the SAPIEN 3 device at 13 participating European and Canadian sites. The SAPIEN 3 THV clinical investigation enrolled patients at both high and intermediate risk for surgery. This analysis includes only intermediate-risk patients with TF delivered devices. Procedures were performed by operators who were experienced with TAVI and previous generations of balloon-expandable devices.
The objective of the SAPIEN 3 Study was to assess the safety and efficacy of the Edwards SAPIEN 3 THV. The primary endpoint was all-cause mortality at 30 days post index procedure. Secondary endpoints were safety, clinical efficacy and echocardiographic valve performance. Clinical outcomes were defined according to the Valve Academic Research Consortium-2 (VARC-2) definitions16. All primary and secondary outcomes were adjudicated by a clinical events committee.
Neurologic status was assessed using the National Institutes of Health Stroke Scale (NIHSS)17, modified Rankin Scale score18 and the Barthel Index19. All neurological examinations were performed by a team member or neurologist at baseline. The NIHSS was also assessed after the procedure, at hospital discharge and at each follow up-visit. Patients with neurological symptoms were evaluated by a neurologist and had all tests performed at each visit. No patients were lost to follow-up at 30 days.
The study was approved by all local ethics committees at each centre and all patients provided written informed consent.
SAPIEN 3 DEVICE AND PROCEDURE
Features of the SAPIEN 3 valve and delivery system have been previously reported15,20. These include the addition of an external polyethylene terephthalate cuff to enhance external sealing, an improved open cell geometry of the stent frame for a very low delivery profile (14 or 16 Fr), as well as an improved distal flex of the delivery system, with a fine adjustment wheel to provide more control of positioning and coaxiality. Procedures were performed as previously described21. Recommendations on the initial positioning of the prosthesis changed during the study period: at the beginning, the position of the centre marker was recommended to be just below the insertion point of the leaflets, which was progressively moved proximally to the base of the cusps in June 2014. Nonetheless, it was suggested that consideration should be given to the percent of prosthesis oversizing, the location of calcifications, device foreshortening and inflation volume, for the final placement of the centre marker. Although the centre marker might help during positioning, it was recommended that it should not be used as a reference during deployment. Patients received aspirin ≥75 mg prior to the procedure and daily afterwards. Clopidogrel 300 mg was administered within six hours of the procedure and followed by 75 mg daily for one month. No particular recommendations were given regarding the use of percutaneous closure or femoral cut-down, and reasons for the selection of one or other strategy were not collected. In most cases, the selection of percutaneous or surgical closure was based on centre/operator preference rather than a technical issue. Likewise, the selection of general anaesthesia vs. conscious sedation and local anaesthesia mostly relied upon the use of transoesophageal echocardiography for guidance of the procedure and surgical cut-downs, and also depended on centre/operator preference.
ECHOCARDIOGRAPHY, ELECTROCARDIOGRAPHY AND COMPUTED TOMOGRAPHY ANALYSES
Echocardiograms were available at baseline in all patients, at hospital discharge in 97 patients (96.0%), and at 30-day follow-up in 92 patients (91.1%). Echocardiographic data were analysed by an independent core laboratory. Aortic regurgitation (AR) was defined as none, trace, mild, moderate, and severe according to a multiparametric approach including regurgitant volume, regurgitant fraction, and effective regurgitant orifice area.
Pre-procedural three-dimensional imaging of the annulus (multidetector computed tomography [MDCT] or transoesophageal echocardiography) was completed in 93 patients. Sizing guidelines were provided by the manufacturer and based on early experiences with the device. The sizing algorithm used was based on systolic annular area measurements from MDCT with the goal of oversizing the native annulus by 2-10% by area22. The final implanted prosthesis size was decided by the local Heart Team according to echocardiographic and/or MDCT measurements.
Pre- and post-implant electrocardiographic (ECG) recordings were analysed in a core laboratory with attention to existing and new conduction abnormalities, new-onset atrial fibrillation, and new permanent pacemaker (PPM) implantation. The indication for PPM implantation was left to the physicians’ discretion.
STATISTICAL ANALYSIS
Categorical variables are expressed as n (%) and continuous variables as mean±standard deviation or median (minimum, maximum), as appropriate. Survival analysis using Kaplan-Meier estimates was used to assess the primary endpoint and the combined endpoint of disabling stroke and mortality. The standard error and confidence limits were computed using Greenwood’s algorithm. For the primary endpoint, the analytical subset was the valve implant population, defined as patients who left the procedure room with a SAPIEN 3 THV implanted. Paired t-tests were used to analyse continuous outcomes and Fisher’s exact tests were used for categorical variables. A two-sided p-value <0.05 was considered statistically significant. All statistical analyses were performed using the SAS software, version 9.3 (SAS Institute, Cary, NC, USA).
Results
A total of 101 patients were included with a mean age of 84.4±3.8 years; 45.5% were male. The baseline characteristics of the study population are displayed in Table 1. The mean STS-PROM score was 5.2±1.7% and the mean logistic EuroSCORE was 13.2±5.1%.
PROCEDURAL FINDINGS AND RESULTS
All procedures were completed through the transfemoral route and there were no crossovers to alternative approaches. The main procedural findings are shown in Table 2. Conscious sedation/local anaesthesia was used in 55 (54.5%) of patients. No conversions to general anaesthesia occurred. A completely percutaneous procedure was performed in 91 patients (90.1%). A THV size 20 mm was implanted in 1 patient (1.0%), size 23 mm in 46 patients (45.5%), size 26 mm in 32 patients (31.7%) and size 29 mm in 22 patients (21.8%). The mean procedure duration, defined as the total time in the procedure room, was 69.9±29.5 minutes.
Technical success (defined as the absence of procedural mortality and correct positioning of only one valve into the proper anatomical location) was achieved in 99 patients (98.0%). One patient (1.0%) required a transcatheter valve-in-valve implantation. Conversion to open heart surgery was necessary in one patient (1.0%) due to LV perforation requiring surgical repair. The THV was retained in the deployed position in this patient. Balloon post-dilation was used in four patients (4.0%). No procedural deaths, coronary obstructions, annulus rupture or valve embolisation were reported (Table 2). The median length of hospital stay (excluding pre-procedure days) was seven days (range two to 34 days).
THIRTY-DAY OUTCOMES
The 30-day outcomes are shown in Table 3. At 30-day follow-up, one patient (1.0%) had died from a periprocedural stroke. A total of three patients (3.0%) suffered strokes: all were ischaemic, with two classified as being disabling (2.0%). Kaplan-Meier curves and estimates of all-cause mortality, mortality or disabling stroke, and of stroke are displayed in Figure 1.
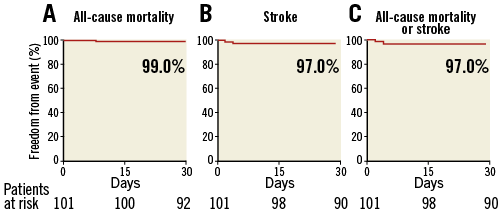
Figure 1. Survival curves after TAVI with the SAPIEN 3 THV. Kaplan-Meier curves showing the percent of patients with all-cause mortality (A), stroke (B) and all-cause mortality or stroke (C) at 30-day follow-up.
A PPM was implanted in four patients (4.0%), 4.3% of patients without pre-procedural PPM. The reason for PPM was third-degree AVB in two patients, second-degree AVB in one patient and atrial flutter/left bundle branch block in the fourth patient. Twenty-two patients (21.8%) had new-onset left bundle branch block. New-onset atrial fibrillation occurred in seven patients (6.9%). Antithrombotic therapy at discharge in patients with pre-existing atrial fibrillation and new-onset atrial fibrillation is shown in Table 4. Two out of seven patients with new-onset atrial fibrillation received anticoagulation therapy. Major vascular complications, life-threatening bleeding and acute kidney injury occurred in two patients (2.0%) each. No congestive heart failure, valve thrombosis or structural valve deterioration requiring repeat procedure was reported. Overall, 12 patients (11.9%) were alive and had experienced no adverse events from implant to 30 days.
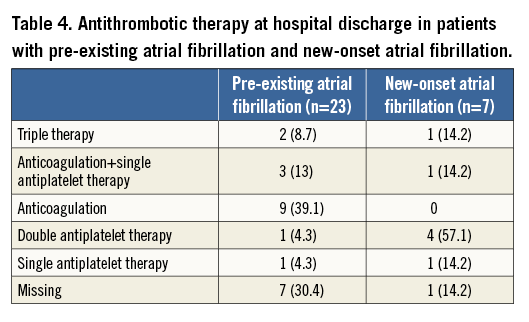
The NYHA functional class and Canadian Cardiovascular Society (CCS) angina scores of the study population at baseline and at 30-day follow-up are displayed in Figure 2A and Figure 2B. A significant improvement was observed in NYHA class after TAVI overall (p-value <0.0001): 75 patients (76.5%) had symptomatic improvement by at least one NYHA functional class and 88.1% (89/101) of patients were in NYHA Class I or II at 30-day follow-up. Significant differences were observed in the CCS angina values before TAVI and at 30-day follow-up (p=0.0001).
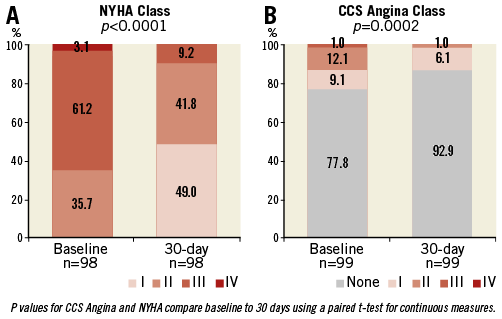
Figure 2. Changes in functional class and angina class. New York Heart Association Class (A) and Canadian Cardiovascular Society Angina Class (B) at baseline and at 30 days after TAVI in the study population.
ECHOCARDIOGRAPHIC RESULTS
Changes in valve haemodynamics are shown in Figure 3. The aortic valve mean gradient significantly decreased in all patients after TAVI procedures (from 47.1±13.3 at baseline to 12.0±4.5 at discharge, p<0.0001). This reduction remained at 30-day follow-up (12.0±4.21 mmHg, p<0.0001 compared to baseline). A significant increase in aortic valve effective orifice area was confirmed at discharge (from 0.7±0.19 to 1.5±0.49 cm², p<0.0001) and remained at 30-day follow-up (1.5±0.40 cm², p<0.0001).
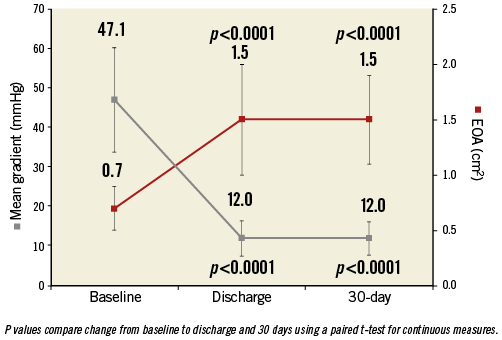
Figure 3. Changes in aortic valve haemodynamics. Aortic valve mean gradient and effective orifice area at baseline, at hospital discharge and at 30-day follow-up. The bars represent standard errors.
No significant changes were observed in left ventricular ejection fraction between baseline (57.3±11.51%, p=0.2596), hospital discharge (54.5±10.72%, p=0.260) and 30-day follow-up (57.8±11.01%, p=0.269).
The incidence and the severity of total aortic regurgitation and paravalvular leaks after TAVI and at 30-day follow-up are shown in Figure 4. No severe paravalvular leaks or total aortic regurgitation were reported during the study period. Mild or more aortic regurgitation occurred in 26 patients (29.2%) with echocardiographic assessments after TAVI without significant changes at 30-day follow-up.
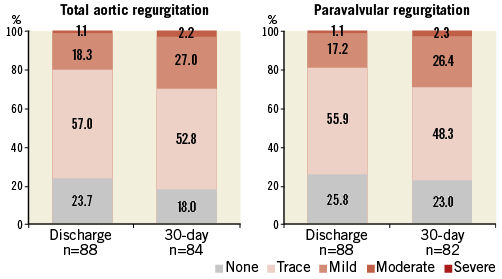
Figure 4. Aortic regurgitation after transcatheter aortic valve implantation. Severity and incidence of total and paravalvular aortic regurgitation at discharge and at 30-day follow-up. A total of nine patients had missing echocardiograms at 30-day follow-up. None of these patients had moderate or severe aortic regurgitation at hospital discharge.
Discussion
Concurrently to a reduction in the rate of complications and the maturity of the technique, a slow but progressive decrease in the risk of patients with an indication for TAVI has occurred in recent years, as revealed by progressively lower surgical risk scores in recent registries8. Several studies have retrospectively evaluated the results of TAVI as compared to surgery in patients with STS or logistic EuroSCORE-defined intermediate risk. Although results varied across studies, a reduced overall 30-day mortality was observed in the TAVI group compared to initial experiences, with no significant differences between TAVI and surgery9,11-13. These studies also suggested a higher survival benefit when these patients underwent TAVI using the transfemoral route12, as has previously been demonstrated for high-risk patients.
The 1.0% mortality observed in this study represents the lowest ever reported in a prospective TAVI trial23-25 and is lower than that previously reported after surgical aortic valve replacement in octogenarian patients24,26,27. The lower-risk profile of patients23, operator experience and the use of the improved SAPIEN 3 THV may explain this very low mortality.
Initial results with the SAPIEN 3 THV showed a reduction in periprocedural complications and mortality in high surgical risk patients consistent with these results, even though these studies represent the initial experience with this device15,20,28. This reduction in mortality corresponded with a reduction in the rate of TAVI-related complications such as vascular complications, bleeding (which were reduced to 2.0%), valve malposition, valve embolisation, annulus rupture and coronary obstruction (which did not occur in this study), and the incidence and severity of paravalvular leaks.
The occurrence of paravalvular leak after TAVI remains a concern. A reduction in the risk of paravalvular leak has been reported with new-generation devices ranging from 18 to 50%29-31. In this study, no patients had severe AR after TAVI and only 2.0% had moderate aortic regurgitation. Mild paravalvular leaks were seen in 26.4% of patients at 30 days.
It has been suggested that the transfemoral approach is a predictor for the occurrence of paravalvular leaks when compared to the transapical approach, due to suboptimal coaxiality and improper positioning of the prosthesis32 during deployment. The Commander delivery system of the SAPIEN 3 THV incorporates modified distal catheter flexing and a fine adjustment wheel allowing improved coaxiality and accurate positioning which, along with the sealing properties of its external cuff, reduce the risk of paravalvular leaks. The reduced risk of paravalvular leak lowers the need for more extensive valve oversizing and balloon post-dilatation compared with its predecessor33,34, something which has also contributed to decreasing the risk of other complications such as annulus rupture.
Initial experiences with the SAPIEN 3 THV showed an increased risk for PPM implantation compared to its predecessors15,35. This has been attributed to low positioning and the longer stent frame of the SAPIEN 3. However, in this study, the rate of PPM implantation was 4.3%, which compares with the rate of PPM implantation after the SAPIEN XT device and approximates to that observed after surgical aortic valve replacement36. This favourable difference might be due to several factors:
1. A lower degree of valve oversizing (~10 vs. the ~20% first suggested).
2. A higher placement position of the prosthesis in order to compensate for the longer stent frame. In fact, initially a 70% supra-annular and 30% infra-annular position was recommended. However, the confirmation that higher positions are not associated with an increased risk of paravalvular leaks has led to a more aortic position of the prosthesis (90:10 or 80:20)37 in order to reduce the risk of conduction disturbances. Nonetheless, the depth of implantation of the prosthesis was not measured in this study and these data have not been confirmed.
3. The lower risk score of patients included in the first phase of the SAPIEN 3 trial.
4. A reduced incidence of pre-procedure conduction disturbances may have contributed at least in part to the reduction of this complication. Indeed, patients at higher risk for atrioventricular block after TAVR (such as patients with pre-existing right bundle branch block) were excluded from this study.
Interestingly, completely percutaneous procedures were performed in more than 90.1% of patients and, in more than half, procedures were performed under conscious sedation/local anaesthesia, which might have led to the lower rate of complications associated with general anaesthesia and intubation. Importantly, no complications occurred in 11.9%.
No decrease was observed in the rate of stroke compared to previous studies, despite the decrease in the rate of new-onset atrial fibrillation and the reduction in the rate of post-dilation and the lower surgical risk of patients. This finding underlines the need for development of new strategies to reduce the risk of this complication.
Limitations
The main limitations of this study are as follows. This is a non-randomised study with a small sample size. However, it is a multicentre study, and all primary and secondary endpoints were evaluated by a clinical events adjudication committee and only patients at intermediate risk for surgery were included in this analysis. In addition, this study has extensive exclusion criteria, and therefore conclusions need to be interpreted with caution when making inferences on the general population. The study lacked an angiography core laboratory to assess implantation height. There were missing echocardiography exams during the follow-up period. Nonetheless, echocardiographic, computed tomography and ECGs were read by independent core laboratories. Data regarding aortic annulus/prosthesis size ratio were calculated using transoesophageal echocardiography dimensions, and therefore might not be accurate.
Conclusions
TAVI performed transfemorally with the SAPIEN 3 THV in patients at intermediate risk for surgery and conducted as a minimally invasive procedure in more than 55% of patients is associated with a very high rate of technical success and very low rate of complications and mortality. Although compelling, this study has several limitations and these results need to be confirmed in randomised trials. The PARTNER II (NCT01314313) and SURTAVI (NCT01586910) trials will provide additional evidence to determine whether TAVI can be recommended in patients at intermediate risk for surgery.
| Impact on daily practice This observational trial adds to the evidence suggesting the efficacy and enhanced safety of the SAPIEN 3 prosthesis, which is a second-generation THV device. This technical progress will contribute to enlarging the indications of TAVI towards lower-risk patients, as well as better patient selection, improved procedural performance, better adjunctive medical treatment/accessory devices and accumulation of evidence. |
Appendix. Exclusion criteria of the SAPIEN 3 Study
Non-calcified aortic valve.
Evidence of an acute myocardial infarction ≤1 month (30 days) before the intended treatment (defined as: Q-wave MI, or non-Q-wave MI with total CK elevation of CK-MB ≥twice normal in the presence of MB elevation and/or troponin level elevation [WHO definition]).
Untreated clinically significant coronary artery disease requiring revascularisation.
Aortic valve is a congenital unicuspid or congenital bicuspid valve.
Mixed aortic valve disease (aortic stenosis and aortic regurgitation with predominant aortic regurgitation >3+).
Pre-existing bioprosthetic valve or ring in any position.
Leukopenia (WBC <3,000 cell/μL), acute anaemia (Hgb <9 g/dL), thrombocytopaenia (platelets <50,000 cell/μL).
Hypertrophic cardiomyopathy with or without obstruction (HOCM).
Severe ventricular dysfunction with LVEF <20%.
Echocardiographic evidence of intracardiac mass, thrombus or vegetation.
Active upper GI bleeding within three months (90 days) prior to procedure.
A known contraindication or hypersensitivity to all anticoagulation regimens, or inability to be anticoagulated for the study procedure.
Native aortic annulus size: a) <18 mm or >25 mm as measured by echocardiography, 23 and 26 mm valves; b) >27 mm as measured by echocardiography, 29 mm valve; c) <16 mm as measured by echocardiography, 20 mm valve.
Clinically (by neurologist) or neuroimaging confirmed stroke or transient ischaemic attack (TIA) within three months (90 days) of the procedure.
Renal insufficiency (creatinine >3.0 mg/dL) and/or renal replacement therapy at the time of screening.
Estimated life expectancy <24 months (730 days) due to carcinomas, chronic liver disease, chronic renal disease or chronic end-stage pulmonary disease.
Expectation that patient will not improve despite treatment of aortic stenosis.
Currently participating in an investigational drug or another device study. Note: trials requiring extended follow-up for products that were investigational, but have since become commercially available, are not considered investigational trials.
Active infection or bacterial endocarditis within six months (180 days) of procedure.
Known hypersensitivity to contrast media, chromium, nickel, molybdenum, manganese, copper, silicon, and/or polymeric materials.
Any neurologic disease which severely affects the ability to walk or perform everyday activities.
Senile dementia.
For intermediate-risk patients enrolled under inclusion criterion 1.c) patients with untreated pre-existing conduction disturbances (AV Block and Bundle Branch Block)
Funding
This trial was sponsored and funded by Edwards Lifesciences, LLC, Irvine, CA, USA.
Conflict of interest statement
A. Vahanian serves on the advisory board of Medtronic and has received speaker’s fees from Edwards Lifesciences. T. Walther has received speaker’s fees from Edwards Lifesciences, St. Jude Medical, and Symetis. O. Wendler is a proctor for the Edwards Lifesciences Transcatheter Heart Valve program. T. Lefèvre is a proctor for transfemoral-TAVI for Edwards Lifesciences and is a consultant for Symetis and Direct Flow Medical. J. Rodés-Cabau is a consultant to and has received a research grant from Edwards Lifesciences. J. Webb is a consultant to Edwards Lifesciences. The other authors have no conflicts of interest to declare.
References
