
Abstract
Aims: Reducing elevated left atrial pressure with an atrial septum shunt device is a possible treatment option in symptomatic heart failure patients. This study aimed to investigate the safety and feasibility of the Atrial Flow Regulator (AFR) in heart failure patients.
Methods and results: AFR-PRELIEVE is a prospective, non-randomised, open-label, multicentre study in patients with symptomatic heart failure NYHA Class III or IV and pulmonary capillary wedge pressure (PCWP) ≥15 mmHg at rest or ≥25 mmHg at exercise irrespective of left ventricular ejection fraction (EF ≥15%). Here we report on procedural and three-month follow-up data for a total of thirty-six enrolled patients. Sixteen (44.5%) patients with reduced EF (HFrEF: EF 15-39%) and twenty (55.5%) patients with preserved EF (HFpEF: EF ≥40%) were enrolled. Implantation success rate and device patency with left-right shunt was 100% (post procedure and at three months) in both patient groups, with one SADE in the HFpEF group which completely resolved. Three (3/36, 8.3%) patients were hospitalised for worsening of heart failure (two HFrEF patients, one HFpEF patient). Individual patients from both the HFrEF and HFpEF groups showed improvement in symptoms and surrogate parameters of heart failure (NYHA class, six-minute walking distance, Kansas City Cardiomyopathy Questionnaire, PCWP, NT-proBNP).
Conclusions: Implantation of the AFR device in heart failure patients is feasible and safe; shunt patency at three months was confirmed in the study. The atrial shunt improved symptoms and surrogate parameters of heart failure in some but not all patients in both the HFpEF and HFrEF groups.
Introduction
Increased left atrial pressure (LAP) leads to exercise intolerance and exertional dyspnoea, and predicts mortality in patients with heart failure (HF)1. Morbidity and mortality rates in HF patients with preserved ejection fraction (HFpEF) are similar to those in HF patients with reduced EF (HFrEF)2,3.
Diastolic dysfunction involves several different haemodynamic and molecular mechanisms, leading to impaired left ventricular (LV) relaxation, development of left atrial (LA) volume overload and pulmonary venous congestion2.
Evidence-based treatments in patients with HFrEF have improved their prognosis4; however, the role of dedicated approaches to reduce elevated LV filling pressure remains unclear. Reducing LAP and LA volume overload using an atrial septum shunt device has emerged as a novel treatment option to improve HF symptoms. Two different devices have been clinically investigated. Implantation of the interatrial shunt device (IASD), tested in HFpEF patients, was proven to be safe and associated with lower pulmonary capillary wedge pressure (PCWP) in a pilot trial, an open-label Phase I and a prospective Phase II trial5,6,7. The first-in-man study of the V-Wave device (V-Wave Ltd, Caesarea, Israel), with an incorporated V-trileaflet porcine tissue valve, demonstrated initial safety and early beneficial clinical and haemodynamic outcomes in patients with HFrEF, though the benefits appeared to be compromised by impaired shunt patency in a single-arm, open-label study8,9.
The present open-label, prospective pilot study (the AFR-PRELIEVE trial) investigated the safety of the Atrial Flow Regulator (AFR; Occlutech, Helsingborg, Sweden) in patients with HF and elevated filling pressures. Herein we present the procedural details, periprocedural and safety events, as well as device patency including three-month results.
Methods
STUDY DESIGN AND POPULATION
AFR-PRELIEVE is a prospective, non-randomised, open-label, multicentre pilot study. Patients were recruited between November 2017 and December 2018 in 10 clinical sites (Germany, Turkey and Belgium). The study was reviewed and approved by the local and national ethics committees before study initiation according to local and national regulations. The study was performed according to current standards including a clinical events committee. Investigators entered all relevant patient information in a dedicated electronic case report form (eCRF). A data safety monitoring board (DSMB) was established. The study was monitored; each site was visited at least once. The funding source as well as the authors analysed the data; the funding source locked the database after final monitoring. The authors of the manuscript had full access to all data. This pilot study has been started on the background of the vast clinical experience with the Occlutech® patent foramen ovale (PFO) and atrial septal defect (ASD) occluder devices, as well as a limited number of compassionate use cases for this particular shunt device implanted in patients with pulmonary hypertension creating a right-to-left shunt.
Inclusion and exclusion criteria are summarised in Table 1. Eligible patients with signed informed consent were approved for the study and underwent right heart catheterisation on the day of the implantation for haemodynamic measurements. Patients who met the haemodynamic study criteria received the Occlutech AFR device. Patients with symptomatic HF (HFpEF or HFrEF) were enrolled consecutively without initial stratification.
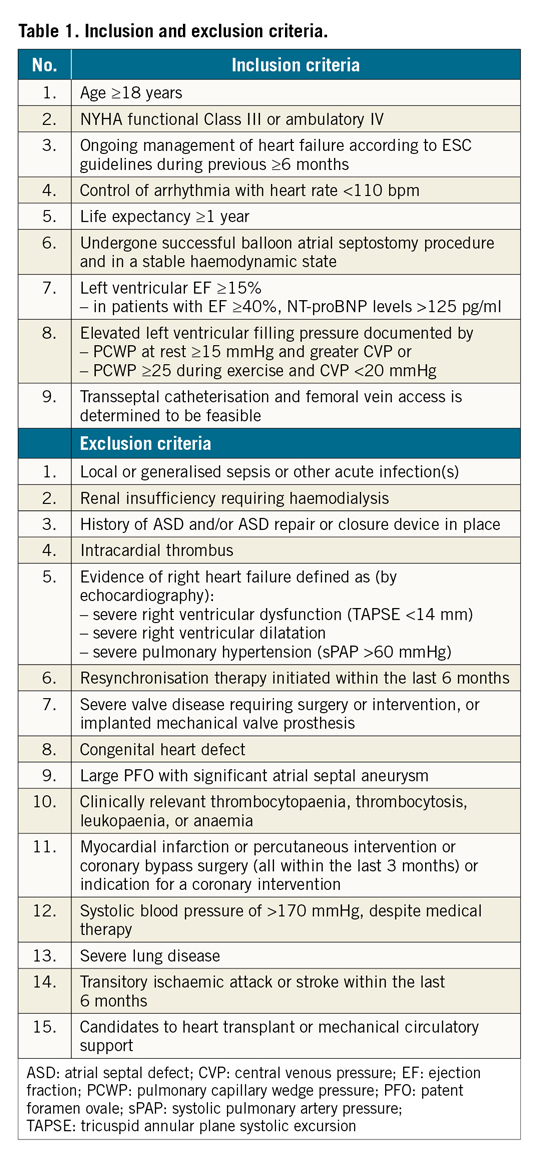
Patients are followed for 12 months (eight clinical visits) to evaluate safety and outcome data of the procedure. NYHA class, six-minute walking distance (6MWD), quality of life assessed by Kansas City Cardiomyopathy Questionnaire (KCCQ) and transthoracic echocardiography parameters are assessed during follow-up according to the protocol, as presented in Figure 1.
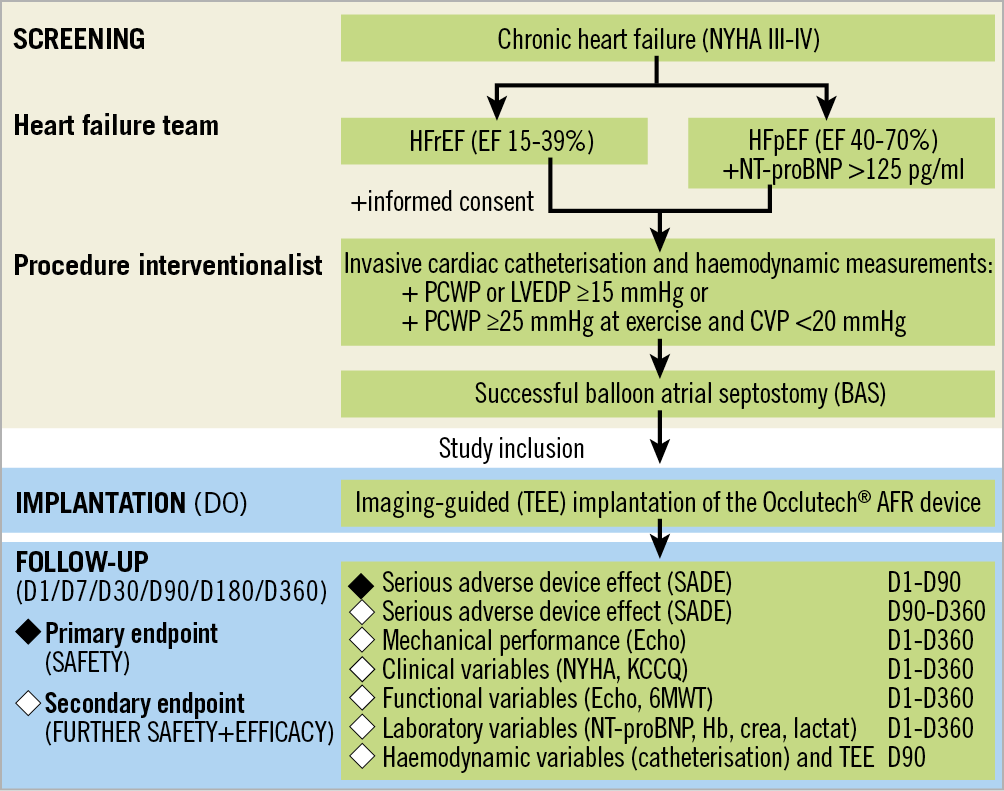
Figure 1. Flow chart of the AFR-PRELIEVE trial.
Transoesophageal echocardiography (TEE) and a right heart catheterisation were pre-specified to be performed at three-month follow-up. Variables measured by echocardiography (echo) are sent to the central reading office for blinded independent validation (coreLab Black Forest GmbH, Bad Krozingen, Germany). However, the reported values are eCRF entries carried out by the local investigators. Each site had to submit a validation echo at the beginning of the study for eligibility.
The primary safety endpoint is the rate of serious adverse device-associated effects (SADEs), assessed at three months and defined as device dislocation/embolisation, damage to the tricuspid or mitral valve caused by the device, intractable arrhythmias caused by the device and any circumstance that requires device removal. Secondary endpoints are the rate of all serious adverse events (SAEs) and further clinical efficacy variables.
PROCEDURE AND AFR DEVICE DESCRIPTION
Before enrolling patients into the study, all interventional cardiology investigators and associated investigative staff at each site underwent training on AFR device implantation and the required medical assessments. Only interventional cardiologists with experience in interventional transcatheter techniques for placement of a PFO or ASD occluder device performed the implantation.
On the day of implantation, a right heart catheterisation was performed to obtain haemodynamic data and provide confirmation of study eligibility. Right atrial, left atrial, pulmonary artery, left ventricular end-diastolic and aortic (systolic and diastolic) pressures, as well as PCWP and cardiac output at rest were measured. PCWP during exercise was measured only in patients who did not reach PCWP ≥15 mmHg at rest. Sizing of the AFR device was performed prior to implantation after careful review of the haemodynamic and anatomical parameters and according to the device sizing instructions (Figure 2). Results from computational predicted haemodynamic pressures in a real-time model of the cardiovascular system to simulate pressure effects at rest and during exercise with an interatrial shunt up to 12 mm led to the selection of the two current AFR sizes of 8 and 10 mm inner fenestration10. As expected, shunt flow increased with increasing shunt size at rest and during exercise, but reached a plateau at a shunt diameter of 10 mm. Furthermore, decrease in PCWP and increase in right atrial pressure also reached a plateau at approximately 9 mm shunt diameter10.
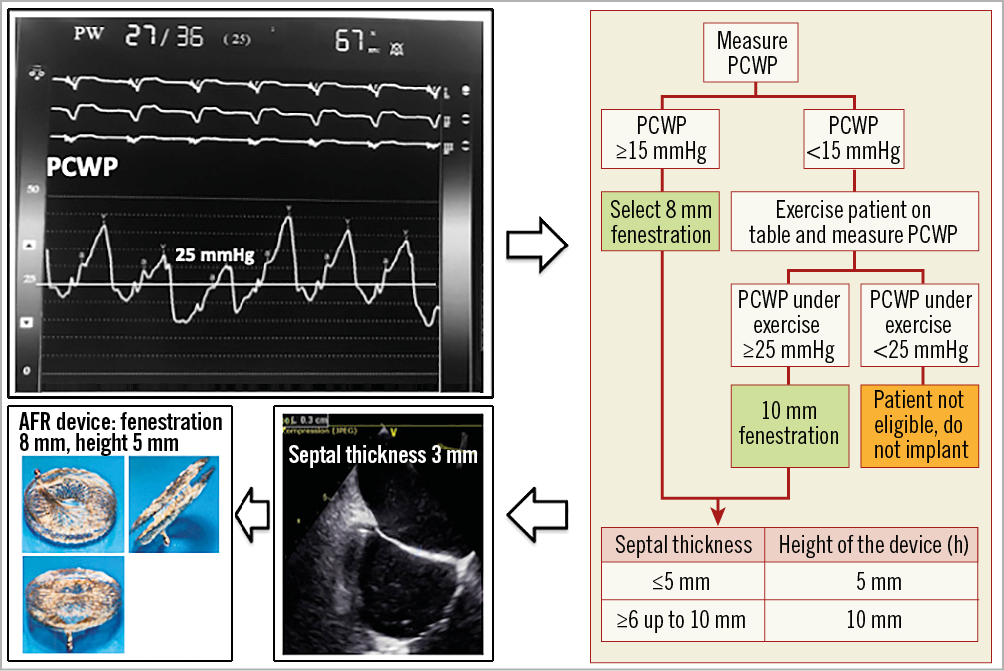
Figure 2. Examples of haemodynamic and echocardiographic measurements with sizing instructions.
Patients were sedated and underwent standard TEE. Examples of intraprocedural images are shown in Figure 3. Following transseptal puncture, a stiff wire was placed into the upper left pulmonary vein. A balloon-based atrial septostomy was performed in all patients; the balloon had a diameter 6 mm larger than the planned fenestration diameter of the AFR device (usually 14 mm). The Occlutech delivery sheath (12 or 14 Fr depending on the AFR device size) was inserted together with a dilatator across the septum into the pulmonary vein over the stiff wire. The Occlutech AFR is a double-disc, circular device made of self-expanding, nitinol wire mesh. A flexible waist in the centre connects the two discs and has a centrally located shunt. A welded ball structure located on the right atrial disc serves as an anchor for the pusher (Flex-II Pusher; Occlutech). After the AFR device is loaded onto the pusher grasps and retracted into the loader, the safety screwing knob secures loading of the device preventing accidental device release. The whole system is advanced through the delivery sheath into the left atrium. Following positioning of the AFR in the LA, the left atrial disc is deployed and positioned at the left side of the septum, similar to PFO or ASD closure devices. Next, the right atrial disc is deployed under constant pull and the correct left/right positioning of the device is confirmed by TEE and angiography. A push and pull manoeuvre confirms stability of the device prior to activation of the release mechanism. Release is performed by unscrewing the security knob at the handle and deployment of the device. Device patency with left-right shunt was documented by TEE after every implantation.
Figure 3. Examples of implantation procedure images of the AFR. Echocardiographic (A) and fluoroscopic (B) images of the transseptal puncture. Echocardiographic (C) and fluoroscopic (D) images of the balloon atrial septostomy (yellow arrow shows the hourglass balloon formation at the start of the septostomy). Echocardiographic (E) and fluoroscopic (F) images of the deployment of the left atrial disc, of the right atrial disc (yellow arrow shows the pull manoeuvre to prove stability before deployment) (H & I), and after release (J & K). Instructions to grip and lock the AFR device on the pusher (G) and for release by opening the locking mechanism (L).
STATISTICAL ANALYSIS
Statistical analysis was performed using GraphPad Prism, version 8 (GraphPad Software, Inc., San Diego, CA, USA). We analysed changes in continuous variables from baseline to three-month follow-up using the paired t-test or Wilcoxon signed-rank test, where appropriate. Haemodynamic and clinical variables were analysed by paired comparison of follow-up versus baseline on an individual patient level. We compared categorical data with Fisher’s exact test. A p-value <0.05 was considered to be statistically significant. Results are reported as mean±standard deviation (SD). NYHA class, 6MWD and KCCQ score analysis results are reported as mean±standard error of the mean (SEM).
Results
ENROLMENT AND BASELINE CHARACTERISTICS
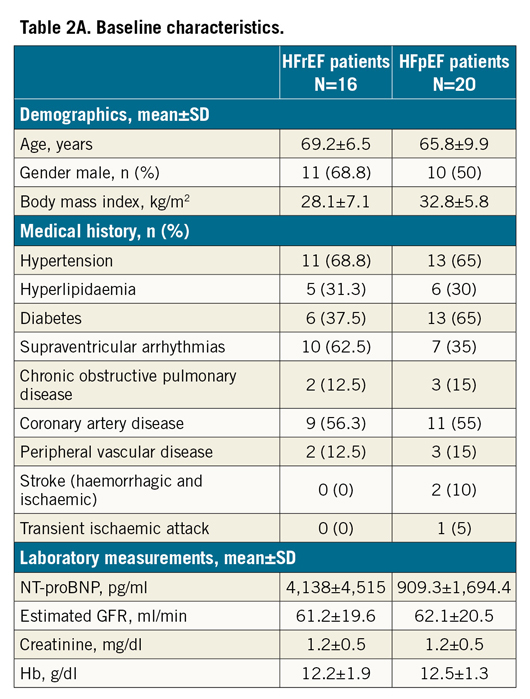
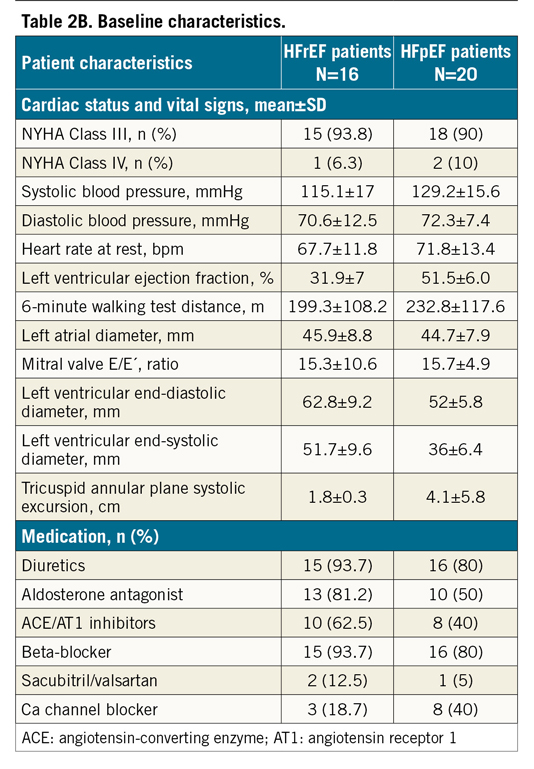
The patient disposition flow chart is presented in Figure 4. Baseline characteristics determined the patient stratification in the two groups (HFrEF and HFpEF) and are summarised in Table 2A and Table 2B. Both HF groups had multiple comorbidities and all participants were on maximal tolerable HF medication at baseline.
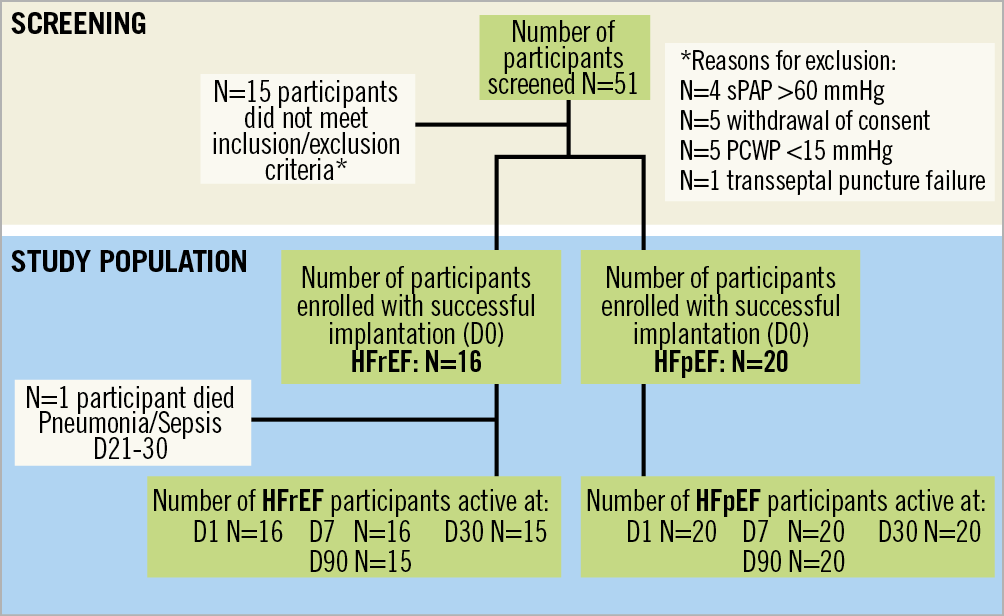
Figure 4. Study participant disposition flow chart of the AFR-PRELIEVE trial.
PROCEDURAL RESULTS
Procedural details are summarised in Table 3. The device has less radial force compared to other devices in the field; therefore, as the stiffness of the septum varies by patient, balloon atrial septostomy is recommended to secure sufficient lumen gain. In this study, as well as previously in the compassionate use cases, there were no issues related to the balloon septostomy.
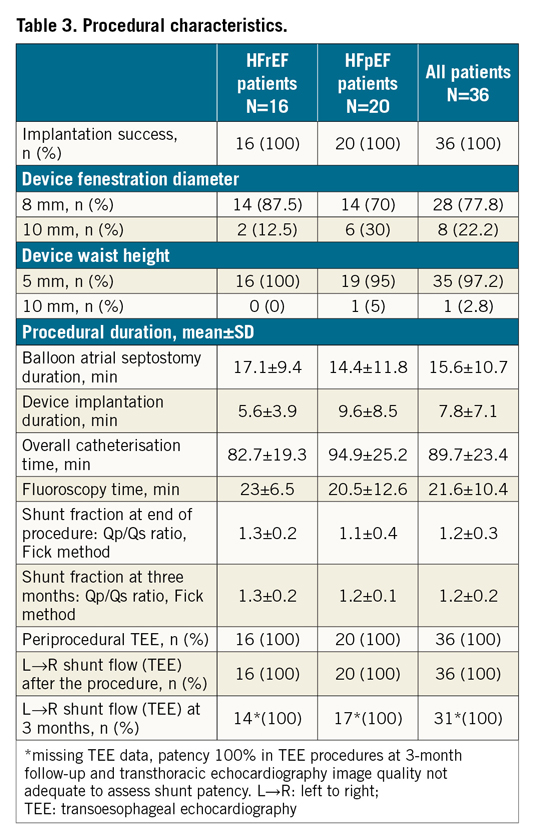
Implantation of the AFR device was successful in all patients with only one placement attempt. In the majority of procedures (HFrEF: 87.5% and HFpEF: 70%), a device with an inner fenestration diameter of 8 mm and a device height of 5 mm (HFrEF: 100% vs. HFpEF: 95% patients) was implanted. Once the device was deployed, the final sizing of the opening was performed with TEE. Depending on the inner fenestration diameter of the device, we measured 8-10 mm without recoil. Left-right shunt was documented using TEE after implantation in all patients. Furthermore, we calculated post-procedural pulmonary to systemic flow ratio (Qp:Qs) to determine the left to right shunt, based on the Fick principle. To provide accurate information, measurements were performed, without providing oxygen-enriched gas. The Qp:Qs ratio was 1.3±0.2, 1.1±0.4 and 1.2±0.3 in the HFrEF, HFpEF and total patient collective, respectively. Both TEE and haemodynamic data calculation confirmed patency of the device at three-month follow-up (Table 3).
PATIENT FOLLOW-UP TO 90 DAYS
A summary of all safety events up to three months is shown in Table 4. In one patient with HFpEF, a procedure-related SAE was documented, i.e., inguinal bleeding after the procedure, no transfusion or surgical intervention needed. One SADE was reported in the same patient, with temporary post-procedural disturbance in consciousness, which was considered possibly related to the study device. No action was taken, and the event resolved without further sequelae. No strokes/TIAs or myocardial infarctions were reported after implantation. One patient (1/36, 2.7%) with HFrEF died 30 days after implantation, due to pneumonia with septicaemia. Three patients (3/36, 8.3%) were hospitalised for worsening of HF. SAE rates up to three months were low in both groups. Five patients (5/36, 13%) had adverse device events, all in the HFpEF group: catheter site reaction, oedema, anaemia, supraventricular arrhythmias and an abnormal respiratory gas exchange were observed during three months of follow-up. All events resolved. No adverse device events were reported in the HFrEF group.
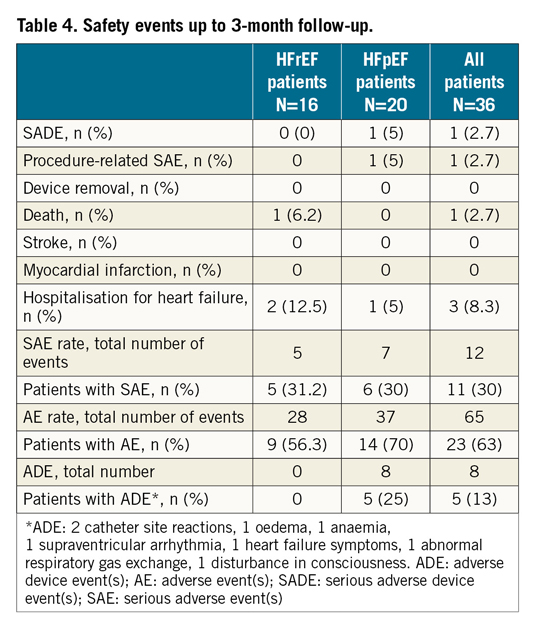
Clinical and haemodynamic variables (paired analysis, follow-up vs baseline, on individual patient level) are depicted in Table 5 and Figure 5A-Figure 5C. Patients improved partially; interpretation of single statistically significant results such as change in NYHA class should be cautious since the patient numbers are small.
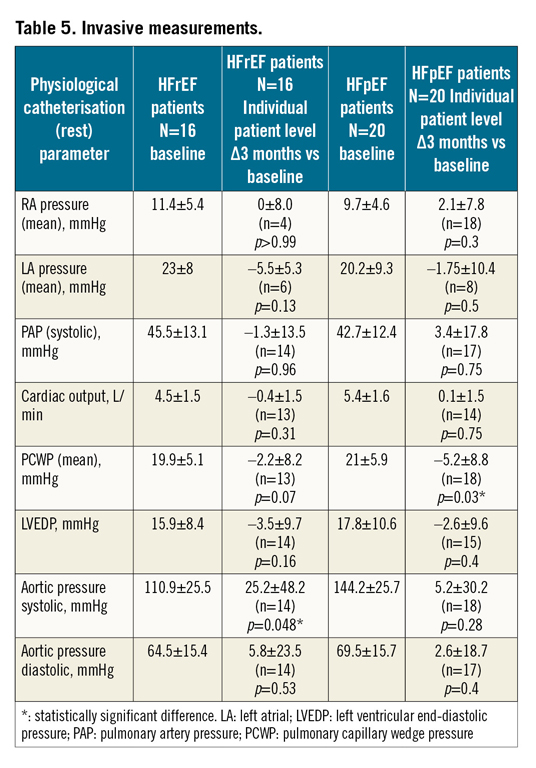
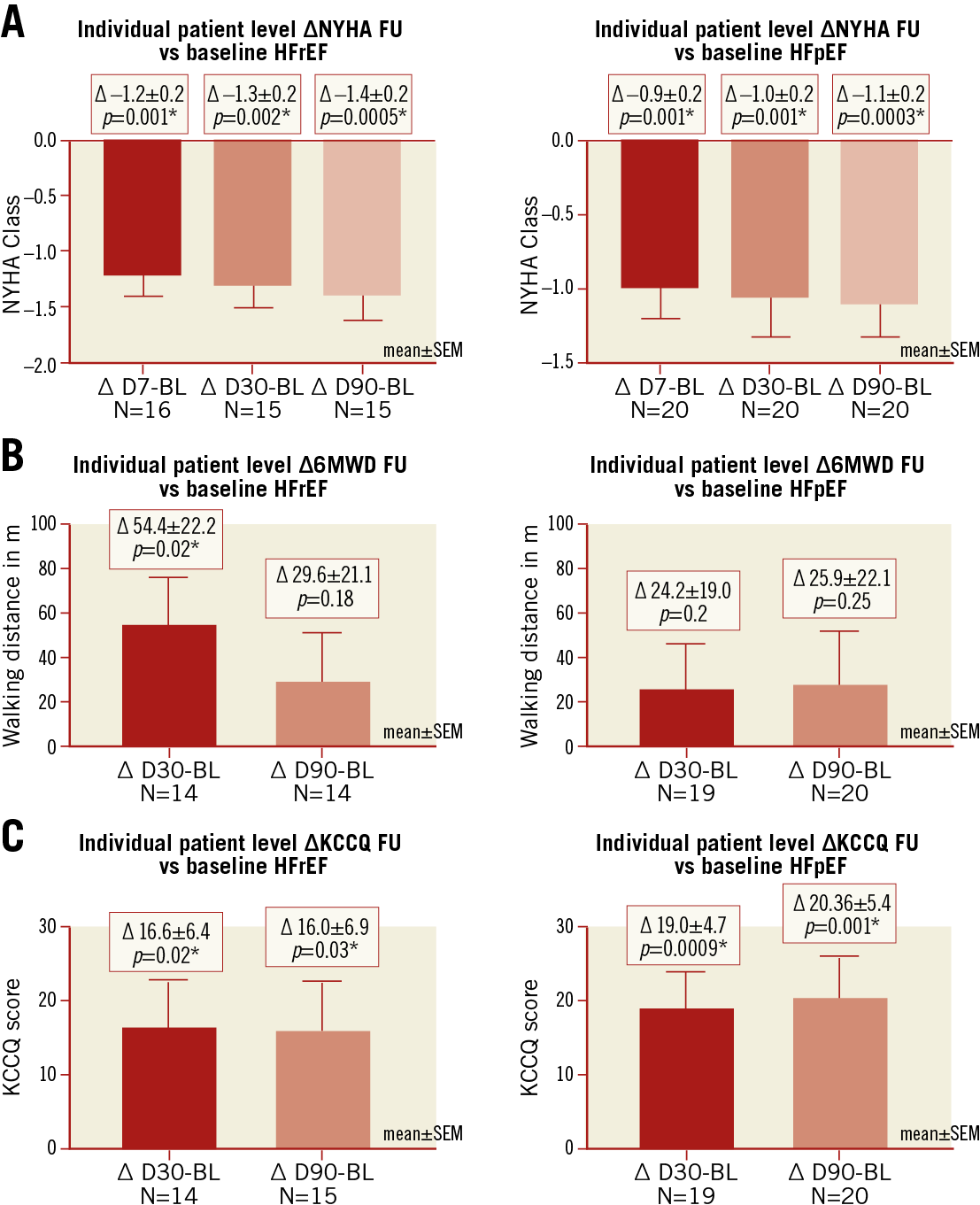
Figure 5. Secondary clinical efficacy endpoint analysis. A) NYHA class. B) Six-minute walking distance (6MWD). C) Kansas City Cardiomyopathy Questionnaire (KCCQ) score. *p<0.05: significant change.
Discussion
AFR-PRELIEVE is a prospective, non-randomised, multicentre Phase II pilot study aiming to assess the safety and feasibility of the Atrial Flow Regulator (AFR) in symptomatic HFrEF and HFpEF patients with elevated PCWP ≥15 mmHg at rest or ≥25 mmHg at exercise. Here, we report the procedural and three-month results. Implantation of the AFR was feasible and safe in all patients. The shunts remained patent at three months.
Diastolic dysfunction with impaired LV relaxation and increased LV stiffness leads to elevated LV filling pressures, atrial volume overload and pulmonary congestion causing dyspnoea symptoms3. It is observed in patients with HFpEF and HFrEF11. According to the “single syndrome” hypothesis of HF, diastolic LV dysfunction has a similar mechanism across the entire HF spectrum12,13,14. An excessive rise of PCWP during exercise, despite normal PCWP at rest in patients with HFpEF, is associated with increased mortality1. In patients with HFrEF, diastolic dysfunction remains impaired despite adequate medical therapy and is highly predictive of worse outcome11,15. The pilot study included patients with elevated LV filling pressures irrespective of the EF, which led to the formation of two patient collectives: sixteen patients with HFrEF (EF 15-39%) and twenty patients with HFpEF (EF ≥40%).
Guideline-recommended drug treatment in patients with HFrEF can improve their outcome; however, current treatment strategies have failed to reduce morbidity or mortality convincingly in patients with HFpEF16. The lack of efficient treatment options in HFpEF prompted the evaluation of new device-based approaches. Device-based reduction of increased LAP is under investigation in patients with symptomatic HF and high filling pressures. The presence of an ASD in patients with mitral valve stenosis, known as Lutembacher syndrome, is associated with fewer symptoms and improved outcomes compared to pure mitral stenosis17. Furthermore, progression to HF following ASD closure has been observed in adult patients and is characterised by acute pulmonary congestion, which is manifested by acute atrial volume overload18. Continuous, invasive measurement of LAP to guide medical therapy in patients with HFrEF was associated with reduced LAP and improved symptoms19.
Currently, there are two devices under clinical investigation, namely the InterAtrial Shunt Device (IASD®; Corvia Medical, Tewksbury, MA, USA), and the V-Wave device6,7,8,9. The AFR device differs from these devices in several aspects. It has no incorporated valve tissue. The interatrial communication is larger than the V-Wave (5 mm) and equal to or larger than the IASD (8 mm). The AFR device is uncoated, made of nitinol mesh. A procedural difference is the necessity to perform a balloon atrial septostomy prior to AFR implantation, which was performed safely in all cases. Procedural success rate and patency of the device (confirmed by TEE post procedure and at three-month follow-up) was 100%. The implantation procedure is relatively similar to the placement of an ASD closure device with a relatively short device implantation time (<10 min). The embolisation risk of the device during the procedure is low, because it is fully retrievable up to final deployment. No stroke/TIA or thrombus on the device was observed at three months using TEE.
One hundred patients have been treated worldwide with the AFR device as compassionate use for pulmonary arterial hypertension, severe heart failure and congenital heart disease, mostly to create a right-to-left shunt20.
Outcome and long-term effects of LA decompression remain incompletely understood. In line with the pathophysiology of diastolic HF, the creation of a controlled left-right shunt may reduce LAP and improve HF symptoms, although a chronic left-right shunt may hypothetically increase the risk of right heart failure. Of note, adults with smaller ASDs (diameter <10 mm and Qp:Qs ratio <1.5) are usually not haemodynamically compromised and seldom develop right heart dilatation or failure21. The three-month haemodynamic evaluation in this pilot study showed no significant increase in pulmonary artery pressure (PAP).
Early evaluation of clinical efficacy up to three months post procedure suggests symptom reduction in individual patients. Indeed, at three-month follow-up, improvement in NYHA class and quality of life (KCCQ) as well as 6MWD was observed in some patients of both collectives. Future studies will need to determine parameters that allow identification of patients who will benefit from this novel approach.
Study limitations
This study is limited by its small sample size, the open-label, non-randomised nature and the absence of a control group. Follow-up is limited to three months post procedure in this report, but the pilot study is ongoing. The study is expected to provide continued insights with additional data collection and analysis. We analysed data separately for HFrEF and HFpEF patients, because the broad inclusion criteria led to clinical differences in the enrolled patient collective. Some secondary clinical outcome parameters are obtained through subjective tests (NYHA class, quality of life) by unblinded participants and investigators; however, additional endpoints not subject to bias (haemodynamic and laboratory measurements, echocardiographic parameters evaluated by a blinded central core lab) are also being investigated.
Conclusions
Procedural and three-month follow-up data indicate that implantation of the AFR device is feasible and safe. Individual patients show improved symptom control and surrogate parameters of heart failure.
|
Impact on daily practice HFrEF patients have a poor prognosis even with currently available guideline-recommended therapy. So far, no effective treatment for HFpEF has been identified. Reducing LAP and LA volume overload with a percutaneously delivered atrial septum device is a novel therapeutic approach for HF patients with elevated filling pressures. The first report of the AFR-PRELIEVE pilot study indicates that the implantation of the AFR shunt device is feasible and safe for HF patients. |
Funding
The study was funded by Occlutech S.A.
Conflict of interest statement
M.W. Bergmann has received lecture fees from Occlutech. The other authors have no conflicts of interest to declare.
