Abstract
Aims: The safety and performance of the Absorb Bioresorbable Vascular Scaffold (Absorb BVS) system (Abbott Vascular, Santa Clara, CA, USA) has been previously established in 131 patients from cohort A and cohort B of the first-in-man ABSORB trial. Following this trial, ABSORB EXTEND was initiated as a global continued access study (outside of the USA) to expand experience with the Absorb BVS system to different geographies with broader inclusion criteria to include the treatment of longer lesions and multiple vessels. We report in this manuscript the twelve-month clinical outcomes of the first 512 patients in this population.
Methods and results: ABSORB EXTEND is a prospective, single-arm, open-label clinical study which will enrol up to 800 patients at up to 100 sites. Included are patients with lesions ≤28 mm in length and reference vessel diameter of 2.0-3.8 mm (as assessed by on-line QCA or IVUS). Treatment of a maximum of two de novo native coronary artery lesions is permitted when each lesion is located in a different epicardial vessel. An independent clinical events committee adjudicates all endpoint-related events. At one year, for the first 512 patients enrolled in the study, the composite endpoints of ischaemia-driven MACE and ischaemia-driven target vessel failure were 4.3% and 4.9%, respectively. The cumulative rate of ARC defined definite and probable scaffold thrombosis for this population was 0.8% at one year.
Conclusions: This interim analysis of the ABSORB EXTEND study shows low rates of MACE and scaffold thrombosis. The study is registered on clinicaltrials.gov (unique identifier NCT01023789).
Abbreviations
ARC: Academic Research Consortium
BVS: bioresorbable vascular scaffold
CEC: clinical events committee
DAPT : dual antiplatelet therapy
DES: drug-eluting stent
DSMB: Data Safety Monitoring Board
EES: everolimus-eluting stent
ID: ischaemia-driven
IVUS: intravascular ultrasound
MACE: major adverse cardiac events (cardiac death/all MI/ID-TLR)
MI: myocardial infarction
PCI: percutaneous coronary intervention
QCA: quantitative coronary angiography
ST: scaffold thrombosis
TVF: target vessel failure (cardiac death/all MI/ID-TLR/ID-Non-TL-TVR)
TLR: target lesion revascularisation
TVR: target vessel revascularisation
Introduction
The advent of drug-eluting stents as a treatment strategy for percutaneous coronary intervention has greatly improved outcomes by addressing the problem of excessive neointimal growth through the addition of antiproliferative agents coated on the stent surface. However, the permanent presence of metallic devices and durable polymers inside the coronary artery might preclude the natural healing process of the vessel, resulting in sustained local inflammatory response and untoward clinical outcomes.
Recently, PCI with bioabsorbable vascular scaffolds has created interest because the need for mechanical support for the healing artery is temporary, and beyond the first few months there are potential disadvantages of a permanent metallic prosthesis. Additionally, this novel technology might help to restore normal vessel reactivity1 and allow positive remodelling2, reducing the stimulus for chronic inflammation and facilitating further interventions in the vessel/lesion or side branches either by percutaneous or surgical means.
The Absorb Bioresorbable Vascular Scaffold (Absorb BVS) system is a fully bioresorbable polymeric scaffold designed to revascularise the vessel like a new-generation metallic DES. This is achieved by providing controlled elution of an antiproliferative drug to minimise neointimal growth, followed by benign resorption into the body after vessel healing and remodelling are complete. This device was first evaluated in humans in the ABSORB clinical trial (cohorts A and B)3,4 with very promising initial results, especially in cohort B, using a slower degrading version of the device.
Based on the results of the ABSORB trial, ABSORB EXTEND was initiated in order to continue to assess the safety and performance of the Absorb BVS in a larger, more diverse subject population with increased lesion complexity. The current interim report summarises the twelve-month analysis of the first 512 patients from the ABSORB EXTEND data set, and provides an initial assessment of the safety and performance of the Absorb BVS in an expanded patient pool.
Methods
STUDY DESIGN AND POPULATION
The ABSORB EXTEND trial is a prospective, single-arm, open-label clinical study that is planned to enrol up to 800 patients at up to 100 global sites. A subset of up to 50 patients who receive planned overlapping Absorb BVS at selected sites with OCT capability will be designated as the OCT subgroup, and a subset of up to 100 patients who receive at least one Absorb BVS at selected sites with multislice computed tomography (MSCT) capability will be designated as the MSCT subgroup. These substudies will be reported separately.
Patients were eligible if they were ≥18 years old with evidence of myocardial ischaemia (e.g., stable or unstable angina, silent ischaemia, positive functional study or a reversible change in the 12-lead electrocardiogram [ECG] consistent with ischaemia). Target vessels (for patients included in this report) should have reference vessel diameter (RVD) ≥2.0 mm and ≤3.3 mm with a maximum lesion length (LL) of ≤28 mm, a percentage diameter stenosis ≥50% and <100% and a Thrombolysis In Myocardial Infarction (TIMI) flow grade of ≥1.
The study excludes patients who have other medical illnesses (e.g., cancer or congestive heart failure) or known history of substance abuse (alcohol, cocaine, heroin, etc.) that in the judgement of the investigator may cause non-compliance with the clinical investigation plan, confound the data interpretation or are associated with a limited life expectancy (i.e., less than one year). Hence, patients who cannot receive prolonged DAPT cannot be enrolled into the study. In addition, major exclusion criteria include presentation with acute myocardial infarction within three days prior to the index procedure, unstable arrhythmias, left ventricular ejection fraction <30%, renal insufficiency and use of chronic anticoagulation therapy that cannot be stopped. Per the study protocol, patients are also excluded if target lesions are located in the left main or within an arterial or saphenous vein graft, in-stent restenotic, totally occluded (TIMI flow grade 0) prior to wire crossing or have been previously stented or treated with brachytherapy or involve a bifurcation with a side branch ≥2 mm in diameter and or ostial lesion >40% stenosed by visual estimation, or a side branch requiring predilatation. Also excluded are lesions with excessive tortuosity, heavy calcification, or visible thrombus.
This study is sponsored and funded by Abbott Vascular, Santa Clara, CA, USA. The research ethics committee of each participating institution has approved the protocol and all enrolled patients provided written informed consent before inclusion.
STUDY DEVICE
The study device (Abbott Vascular) is the same as used for the ABSORB cohort B trial, and has been described in detail previously5,6. In brief, the balloon-expandable Absorb BVS comprises a poly L-lactide (PLLA) backbone, coated with a matrix composed of the antiproliferative drug everolimus (Novartis Pharmaceuticals Corporation, Basel, Switzerland) and polymer poly (D, L-lactide) (PDLLA) in a 1:1 ratio to form an amorphous drug-eluting coating matrix containing 100 µ everolimus/cm2. Both PLLA and PDLLA are fully bioresorbable; PDLLA is expected to be totally resorbed by the body in nine months and PLLA in approximately 36 months. During the resorption process, ester bonds in the PLLA and PDLLA chains are hydrolysed. The ultimate degradation product of both PLLA and PDLLA is lactic acid, which is biologically ubiquitous and metabolised via the Krebs cycle4.
At the time of the inclusion of this patient set, for the first 512 enrolled patients, scaffolds were available in diameters of 2.5 and 3.0 mm, and lengths of 18 and 28 mm.
STUDY PROCEDURE
Patients were registered through an interactive voice response system (Oracle America, Inc. Woburn, MA, USA) following confirmation of angiographic inclusion criteria and delivery of the Absorb BVS device beyond the guiding catheter. Registered patients are to remain in the study until completion of the required follow-up period.
A maximum of two de novo native coronary artery lesions could be treated, each located in a different major epicardial vessel. The recommended range for target vessel diameter was assessed in terms of the online QCA or IVUS parameters of distal maximum lumen diameter (Dmax) and proximal Dmax, which refer to the maximum lumen diameter evaluated at the distal and proximal ends of the target segment to be scaffolded, respectively. Planned overlapping of scaffolds was permitted in lesions >22 mm and ≤28 mm in length, with an overlap of 1 mm to 4 mm. All target lesions were to be treated using standard interventional techniques with mandatory predilation and scaffold implantation at a pressure not exceeding the rated burst pressure. Post-dilatation was left to the discretion of the investigator; however, if performed, it was to be with a non-compliant balloon sized to fit within the boundaries of the scaffold. An unexpanded scaffold was not allowed to be reintroduced into the artery once pulled back into the guiding catheter. In the event of bail-out and additional stent/scaffold requirement, a commercial EES or Absorb BVS system was recommended.
All patients enrolled in the study were to be pretreated with a loading dose of ≥300 mg of clopidogrel and ≥300 mg of aspirin, followed by 75 mg of clopidogrel daily for a minimum of six months with ≥75 mg of aspirin for a minimum of three years.
Source document verification
Source document verification (SDV) was routinely performed in 100% of all reported events and 100% of patients through 30-day follow-up. Subsequently, SDV was performed in a random 20% of patients for the remaining follow-up visits.
Follow-up
Assessment of anginal status, data collection of adverse events, details of any subsequent coronary interventions, and use and changes in concomitant medications are to be collected at 30 days (±7 days), 180 days (±14 days) and one, two and three years (±28 days).
Study endpoints and definitions
In addition to acute success, which is composed of clinical device (analysed on a per lesion basis) and clinical procedure success (analysed on a per subject basis), endpoints include adjudicated scaffold thrombosis (ST), cardiac death, MI (target and non-target vessel), and revascularisation (TLR/TVR/all revascularisations) rates. The composite rates of ischaemia-driven MACE (ID-MACE), ischaemia-driven target vessel failure (ID-TVF), ischaemia-driven target lesion revascularisation (ID-TLR) and ischaemia-driven target vessel revascularisation (ID-TVR) were also analysed.
All study endpoint events were adjudicated by an independent clinical events committee (CEC) according to either protocol definitions and/or the Academic Research Consortium (ARC) definitions7. All adverse events were reported to an independent data and safety monitoring board (DSMB), which reviewed the data to identify any safety issues related to the conduct of the study.
Clinical device success was defined as successful delivery and deployment of the clinical investigation scaffold at the target lesion and successful withdrawal of the scaffold delivery system with attainment of final residual stenosis <50% by QCA (by visual estimation if QCA is unavailable). Standard predilation catheters and post-dilatation catheters (if applicable) may be used. Bail-out patients will be included as device success only if the above criteria for clinical device success are met.
Clinical procedure success was defined as successful delivery and deployment of the clinical investigation scaffold at the target lesion and successful withdrawal of the scaffold delivery system with attainment of final residual stenosis of <50% by QCA (by visual estimation if QCA unavailable) and/or using any adjunctive device without the occurrence of ischaemia-driven major adverse cardiac events (MACE) during the hospital stay with a maximum of first seven days post index procedure. In a dual lesion setting, both lesions must have met clinical procedure success.
Cardiac death was defined as any death due to proximate cardiac cause (e.g., MI, low-output failure, fatal arrhythmia). Unwitnessed death and death of unknown cause were classified as cardiac death. This included all procedure-related deaths including those related to concomitant treatment.
MI classification and criteria for diagnosis were defined according to the per protocol definition; Q-wave MI was the development of a new, pathological Q-wave. Non-QMI was elevation of CK levels to ≥ two times the upper limit of normal with elevated CK-MB in the absence of new pathological Q-waves.
Revascularisation events were defined as:
– Ischaemia-driven major adverse cardiac event (ID-MACE): composed of cardiac death; myocardial infarction (MI, classified as Q-wave and non-Q-wave MI); ischaemia-driven target lesion revascularisation (TLR) by CABG or PCI.
– Ischaemia-driven target vessel failure (ID-TVF): composed of cardiac death; myocardial infarction (Q-wave and non-Q-wave); ischaemia-driven target vessel revascularisation by CABG or PCI.
– Ischaemia-driven target lesion revascularisation (ID-TLR): ID-TLR was defined as any repeat percutaneous intervention of the target lesion or bypass surgery of the target vessel with either positive functional ischaemia study; ischaemic symptoms and angiographic minimal lumen diameter stenosis ≥50% by core laboratory QCA; or revascularisation of a target lesion with diameter stenosis ≥70% by core laboratory QCA without either ischaemic symptoms or a positive functional study.
– Ischaemia-driven target vessel revascularisation (ID-TVR): ID-TVR was defined as any repeat percutaneous intervention or surgical bypass of any segment of the target vessel with either positive functional ischaemia study; or ischaemic symptoms and an angiographic minimal lumen diameter stenosis ≥50% by core laboratory quantitative coronary angiography (QCA); or revascularisation of a target vessel with diameter stenosis ≥70% by core laboratory QCA without either ischaemic symptoms or a positive functional study.
Scaffold thrombosis was categorised as acute (<1 day), subacute (1-30 days) and late (>30 days) and was defined according to the ARC guidelines as follows: definite: acute coronary syndrome and angiographic or pathologic confirmation of scaffold thrombosis; probable: unexplained death ≤30 days or TV-MI without angiographic information.
Statistical analysis
The sample size for ABSORB EXTEND was not defined on the basis of an endpoint hypothesis but rather to provide moderate precision on device performance. A sample size of 800 subjects will produce a moderately accurate two-sided 95% confidence interval around clinical endpoints estimates. The half width of the two-sided confidence interval around the two-year scaffold thrombosis rate estimate will be between 0.6% and 0.8%, assuming the true rate between 0.5% and 1.0%.
Binary variables are presented as percentages. Mean and standard deviation are presented for continuous variables.
Results
The current report presents an analysis of the first 512 patients from the ABSORB EXTEND data set who were enrolled from 11th January 2010 to 3rd December 2012 at 56 sites in Europe, Australia, New Zealand, India, Japan, Hong Kong, Malaysia, Singapore, Latin America and Canada. Factors influencing rates of patient recruitment included medical ethics committee approval times and product availability. Clinical follow-up data were obtained for each patient through 12 months. Clinical follow-up was achieved for all the enrolled patients.
Patient demographics are shown in Table 1. The mean age was 62 years (±11) and 74% of patients were male. In addition, 26% of patients were diabetic, 41% of lesions were class B2 or C according to the American College of Cardiology-American Heart Association, and 7% of patients had treatment of two target lesions. Unstable angina was present in 31% of patients with 15% Braunwald Class III.
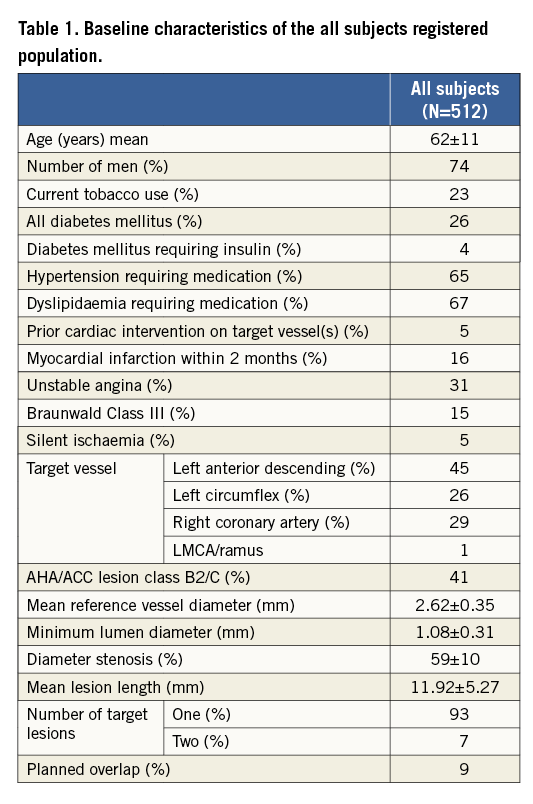
Lesions, as assessed by on-line QCA, had a mean reference vessel diameter of 2.62±0.35 mm and mean lesion length of 11.92 mm (range minimum-maximum 3.13, 33.78). Lesion length was between 10 mm and 20 mm in 48.5% of lesions and planned overlapping scaffold treatment occurred in 8.8%. The majority of treated lesions were in the left anterior descending artery (44.5%). The majority of scaffolds implanted were 3.0 mm×18 mm followed by 3.0 mm×28 mm and 2.5 mm×18 mm.
Clinical device success was 98.5% (540/548) for the 548 lesions treated. An Absorb BVS was not implanted in eight lesions (three of the cases were due to an inability to cross the target lesion, four were due to a failure to reach the target lesion, and one was not implanted due to the scaffold being too small for the lesion), which accounts for the eight failures. The clinical procedural success was 96.9% (496/512) for the 512 patients treated. Aside from the eight patients in whom there was a device failure, an additional eight patients were adjudicated as having a NQMI during the hospital stay within a maximum of the first seven days (which meets the requirement for being a clinical procedural failure). Clinical outcomes through one year are presented in Table 2. At 30 days, the adjudicated composite rates of ID-MACE and ID-TVF were both 2.1% (11/512), driven by eight patients (1.6%) who experienced NQMI during the in-hospital phase and an additional three patients (0.6%) who suffered a QMI within the first 30 days. At 30 days, the non-hierarchical rates of cardiac death, ID-TLR and ID-Non-TLR TVR were 0.0%, 0.2% and 0.0%, respectively.
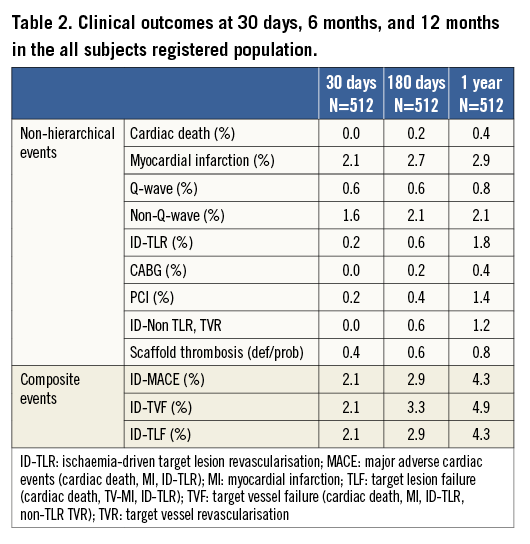
At six months the composite rates of ID-MACE and ID-TVF were 2.9% (15/512) and 3.3% (17/512), respectively, with a single cardiac death (0.2%), while the MI rate was 2.7%. No additional QMI occurred between 30 days and six months; however, there were three additional patients who experienced a NQMI (2.1%). The non-hierarchical ID-TLR rate within the first six months of clinical follow-up was 0.6% (3/512) with one patient undergoing a CABG and one patient a PCI revascularisation between 30 days and six months. In addition, by six months, three patients had undergone an ID-Non-TLR TVR, one by PCI and two by CABG.
At one year, the ID-MACE and ID-TVF rates remained very low, at 4.3% (22/512) and 4.9% (25/512), respectively. One additional QMI and six ID-TLR, five by PCI and one by CABG, were reported between six and 12 months. Three additional patients underwent an ID-NTL TVR (all three by PCI). There was one additional cardiac death reported.
The ARC-defined acute, subacute and late scaffold thrombosis (ST) (definite and probable) rates were 0.0%, 0.4% and 0.4% (0/512; 2/512; 2/512), respectively. The four cases have been previously reported8.
At one year, 83.6% of patients were taking clopidogrel, ticlopidine or prasugrel and 96.1% of patients were taking aspirin, with 81.3% of patients on DAPT.
Discussion
Many post-market single-arm studies are currently ongoing to assess the efficacy of the Absorb BVS in complex lesions or in patients who would otherwise be excluded from the ABSORB EXTEND registry. Therefore, the authors have presented data from the first 512 patients enrolled in this registry to ensure timely publication and ensure the relevance of these data on simple lesions not representative of daily real-world practice.
This interim analysis of the ABSORB EXTEND registry confirms, in a larger and more complex population, the efficacy of the Absorb BVS, with very low occurrence of ischaemia-driven target vessel failure and scaffold thrombosis events.
The ABSORB clinical programme was initiated with the ABSORB (cohort A and B) trial. Cohort A of the trial enrolled 30 patients and has shown good clinical safety and performance through four-year follow-up9. IVUS results showed significant increases in luminal area and in minimum lumen area from 180 days to two years, a finding not observed in any previous clinical study of conventional balloon-expandable metallic stents10. After minor modifications to the scaffold design and processing, cohort B enrolled 101 patients. Clinical follow-up data from all patients are now available to two years4,11. These data confirmed the outcomes of safety and performance established in the cohort A investigation.
The ABSORB EXTEND study was meant to evaluate the clinical safety of this device in a population with more complex lesion characteristics, in order to enable a more accurate estimate of the MACE and scaffold thrombosis rates associated with Absorb BVS than was possible with the smaller first-in-man trial.
While ABSORB EXTEND is a single-arm study, its results could be interpreted in the context of contemporary clinical trials with metallic DES which had similar inclusion/exclusion criteria. For instance, patients treated with the XIENCE V everolimus-eluting stent (Abbott Vascular) in the SPIRIT II (n=223) and III (n=669) trials had a comparable clinical and angiographic profile and at the end of one year of clinical follow-up presented MACE rate of 2.7% and 6.0% with stent thrombosis rate (by protocol definition) of 0.5% and 0.8%, respectively12,13. In the Resolute FIM trial, which enrolled 139 patients treated with the new-generation zotarolimus-eluting stent, the one-year MACE and stent thrombosis rates were 8.7% and 0.7%, respectively14. Although these numbers do not come from a direct comparison between the devices, the low one-year MACE (4.3%) and definite/probable scaffold thrombosis rate (0.8%) in this ABSORB EXTEND interim report resemble the results achieved with the most widespread used contemporary DES.
The present results also compare favourably with the one-year clinical outcomes of cohort B population from the ABSORB trial, despite the more strict inclusion/exclusion criteria and limited number of participating sites in that study. Although cohort B included only one patient with dual target vessels (1%) and the use of overlapping scaffolds was not allowed, (7% and 9%, respectively with the patients enrolled in the ABSORB EXTEND registry), the one-year MACE rate was slightly higher than in the present report (6.9%).
Of note, whether due to the relatively non-complex characteristics of the enrolled population or to the intrinsic properties of this new device, in the entire clinical programme of the Absorb BVS (cohorts A+B and EXTEND interim analysis, n=581), only four cases of scaffold thrombosis were documented. It is important to mention that the present report includes patients treated in several continents (>50 sites), therefore accurately reflecting worldwide interventional practice.
Limitations of the study
This study was limited by the single-arm nature of the design and the inherent lack of a control arm for direct comparison. Although consecutive eligible patients were to be enrolled, a selection bias cannot be excluded. In addition, data from the first 512 patients of the study are presented in this interim analysis and not the entire patient cohort of approximately 800 patients. Finally, the reported follow-up period (one year) might be insufficient to address other potential benefits of this novel technology.
The final report, with longer-term follow-up from this ABSORB EXTEND study, will confirm the presented results.
Conclusion
The clinical results of this interim analysis of the ABSORB EXTEND registry, showing the good acute performance and sustained mid-term efficacy and safety of this novel device, represent another major step toward the incorporation of vascular restoration therapy to the intervention cardiology practice.
| Impact on daily practice Bioresorbable scaffolds are becoming a more frequently used device in daily clinical practice. The current results of the ongoing ABSORB EXTEND registry are reassuring on the efficacy of these devices in the treatment of patients with moderate-complexity coronary disease in a similar fashion to what has been reported in the initial registries with metallic drug-eluting stents. Furthermore, no major concerns about safety of these new devices have been raised. Additional studies with more complex populations and longer follow-up time are warranted. |
Guest Editor
This paper was Guest Edited by Manel Sabaté, MD, PhD, FESC, Servicio de Cardiología, Clínic Hospital, Barcelona, Spain.
Conflict of interest statement
M. Stuteville, C. Dorange, K. Sudhir and W.-F. Cheong are employees of Abbott Vascular. The other authors have no conflicts of interest to declare. The Guest Editor has no conflicts of interest to declare.
