Abstract
Aims: Multimodality imaging of the first-in-man trial using a fully resorbable everolimus-eluting scaffold (BVS, Abbott Vascular, Santa Clara, CA, USA) demonstrated at two years the bioresorption of the device while preventing restenosis. Nevertheless, the long-term safety and efficacy of this novel therapy remain to be documented.
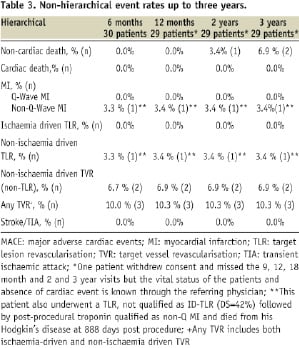
Methods and results: The ABSORB trial completed in July 2006 at four clinical sites in Europe and New Zealand the enrolment of 30 patients with a single de novo native coronary artery lesion. The major clinical endpoint was ischaemia-driven major adverse cardiac events (ID-MACE) defined as a composite of cardiac death, myocardial infarction, or ischaemia-driven target lesion revascularisation. Clinical follow-up was available in 29 patients since one patient withdrew consent. At 46 days, one patient experienced a single episode of chest pain and underwent a diagnostic optical coherence tomography and subsequently a target lesion revascularisation with slight troponin rise after the procedure. At 3-year the hierarchical ID-MACE of 3.4% remained unchanged. Clopidogrel therapy was discontinued in all but one patient. There has been no stent thrombosis reported. Two non-cardiac deaths were reported; one from duodenal perforation, the other from Hodgkin disease. Two patients underwent non-ischaemia driven target vessel revascularisation.
Conclusions: Three-year clinical results have demonstrated a sustained low MACE rate (3.4%) without any late complication such as stent thrombosis.
Introduction
In contrast to bare metal stents (BMS), polymer-based sirolimus-eluting stents (SES) and paclitaxel-eluting stents (PES) have both been shown to significantly reduce angiographic restenosis and recurrent ischaemia necessitating repeat revascularisation1. The occurrence of stent thrombosis, however, remains a clinical concern2,3. Furthermore, permanent metallic stenting may preclude surgical revascularisation, result in jailed side branches, prevent expansive remodelling, eliminate reactive vasomotion and hamper the non-invasive imaging of coronary arteries with either multislice computed tomography (MSCT) or magnetic resonance (MRI). As an alternative approach to the metallic drug-eluting stent, bioresorbable polymer drug-eluting scaffolds may provide short-term vessel scaffolding combined with drug delivery capability without the long-term limitations of metallic stents.
The fully absorbable everolimus-eluting BVS scaffold (Abbott Vascular, Santa Clara, CA, USA) was tested in the first-in-man ABSORB trial enrolling 30 patients with a single de novo coronary artery lesion. Two-year follow-up of the trial with multiple modality imaging can be summarised as follows: 1) bioresorption of polymeric struts (documented with intravascular ultrasonography [IVUS] and optical coherence tomography [OCT], Figure 1); 2) late enlargement of lumen from six months to two years (IVUS and OCT); 3) restoration of vasomotion and endothelial function in some patients; 4) sustained scaffolding of plaque deformability (palpography); 5) feasibility of non-invasive imaging with MSCT; 6) low two-year ischaemia-driven (ID) major adverse cardiac event (MACE) rate of 3.4% without any stent thrombosis.4-9
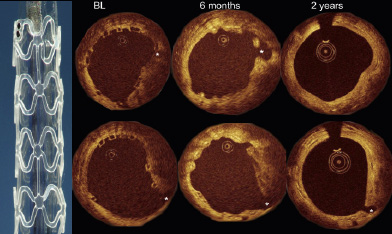
Figure 1. Examples of serial OCT imaging post procedure, at six month and at two years with small side branches (asterisks). Macroscopic view of the BVS is shown in the left side of the illustration. At baseline, the struts were well apposed and recognised as boxes. At six month, the struts were still recognisable with OCT, the luminal contour was irregular with some struts being malapposed (upper row, at 2 o’clock). At two years, the luminal surface was smooth and circular, and some struts were indiscernible. Overall, in quantitative analysis, there was an increase in mean luminal area from six month to two years7.
The long-term outcomes of biodegradation of the BVS scaffold in human coronary arteries however remain to be investigated to confirm the safety and efficacy of the technology. We hereby report the 3-year clinical results in the original cohort of patients treated with a BVS in the ABSORB trial.
Methods
Study design
The design of the current study has been published10. Briefly, in this single-arm, prospective, open-label study, with safety and imaging endpoints, 30 patients were enrolled at four participating sites between March and July 2006. Patients were older than 18 years, with a diagnosis of stable, unstable or silent ischaemia. All treated lesions were single, de novo in a native coronary artery of 3.0 mm, shorter than 8 mm for the 12 mm stent or <14 mm for the 18 mm stent (only two patients received an 18 mm stent), with a percent diameter stenosis ≥50% and <100% with a thrombolysis in myocardial infarction (TIMI) flow grade of ≥1. Major exclusion criteria were patients presenting with an acute myocardial infarction, unstable arrhythmias or patients who had left ventricular ejection fraction <30%, restenotic lesions, lesions located in the left main coronary artery, lesions involving a side branch >2 mm in diameter, and the presence of thrombus or another clinically significant stenosis in the target vessel. A risk analysis of multi-modality imaging based on previously published reports was provided to the local ethics committee11,12. The ethics committees approved the protocol at the participating institutions and the enrolled patients (intention-to-treat [ITT] population) gave written informed consent before inclusion. Four patients were excluded from the per treatment evaluable (PTE) population as they received a non-BVS stent in addition to the study device (BVS). Clinical endpoints were assessed at 30 days, six and nine months, at one, two and three years and will be assessed yearly until five years. Neither invasive (e.g., angiographic) nor non-invasive (e.g., MSCT) imaging tests were planned after two years. All MACE events were adjudicated by an independent Clinical Events Committee and a Data Safety Monitoring Board monitored patient safety.
Study device
The study device, the description of the polymers as well as in vitro and preclinical data has been described elsewhere.10 The polymeric device consists of a backbone of poly-L-lactide (PLLA) coated with poly-D, L-lactide (PDLLA) that contains and controls the release of the anti-proliferative drug, everolimus. The long chains of PLLA and PDLLA are progressively shortened as ester bonds between lactide repeat units are hydrolysed. Ultimately, PLLA and PDLLA fully degrade to lactic acid that is metabolised via the Krebs cycle. In vivo, small particles less than 2 mm in diameter are phagocytosed by macrophages.
Stenting procedure
The BVS scaffold was implanted following mandatory predilatation. Four patients who received a non-BVS stent in addition to a study device were excluded from the per-treatment evaluable population but are included for the clinical endpoints. When additional stenting was required, the Cypher sirolimus-eluting stent (Cordis, Miami, FL, USA) was used since there were no animal/pre-clinical data on overlapping BVS devices available at the time of patient enrolment. All patients were required to receive aspirin ≥75 mg daily for the study duration (five years) and clopidogrel 75 mg daily for a minimum of six months.
Definitions
The composite clinical endpoint is cardiac death, myocardial infarction (MI) classified as Q-wave and non-Q wave MI, and ischaemia driven target lesion revascularisation (TLR). Ischaemia-driven TLR implies revascularisation at the target lesion, irrespective of time window for angiographic control at six months or two years, associated with either i) positive functional ischaemia study, or ii) ischaemic symptoms and angiographic minimal lumen diameter stenosis ≥50% by core laboratory quantitative coronary angiography (QCA), or iii) revascularisation of a target lesion with diameter stenosis ≥70% by core laboratory QCA without either ischaemic symptoms or a positive functional study.
For a diagnosis of non-Q MI, elevation of CK levels ≥2 times the upper limit of normal with elevated CK-MB was required. Stent thrombosis was adjudicated according to the ARC definitions.13 Other clinical endpoints include all-cause mortality, non-ischaemia driven TLR, ischaemia driven or non-ischaemia driven target vessel revascularisation (TVR).
Statistical analysis
For binary variables, percentages are calculated. For continuous variables mean and standard deviation are computed. Unless otherwise noted, the analysis is based on the intention-to-treat population.
Role of funding source
This study was sponsored by Abbott Vascular (Santa Clara, CA, USA). The sponsor was involved in the design and conduct of the study; the collection, management, initial analysis, and interpretation of the data; and had the right to a nonbinding review of the manuscript. Full monitoring ensured accuracy of data collection. The two senior authors (JAO and PWS) had full access to all the data and had final responsibility for the decision to submit for publication. This study is registered with ClinicalTrials.gov, number NCT00300131.
Results
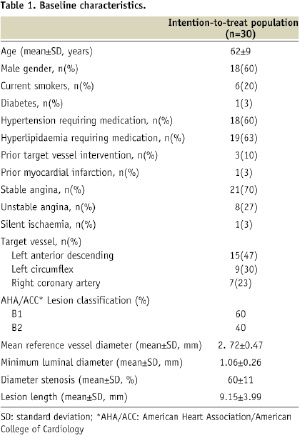
Patient demographics and a flow chart are shown in Table 1 and Figure 2, respectively.
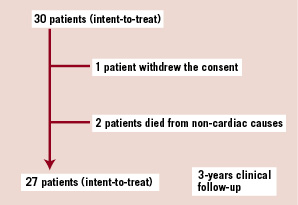
Figure 2. A flow-chart of the patient follow-up is presented. Two patients died from a non-cardiac cause at 706 and 888 days post procedure. One patient withdrew the consent and missed the 9-, 12-, 18- month and 2- and 3- year visits, but his vital and clinical status remained available through his referring physician.
One patient withdrew the consent from the study after the six month follow-up but his vital and clinical status remained available through his referring physicians. Two patients died from non-cardiac cause during follow-up and 3-year clinical follow-up was obtained in the remaining 27 patients. In 29 patients with any available follow-up, the median duration of follow-up was 1,100 days, ranging from 706 to 1,129 days.
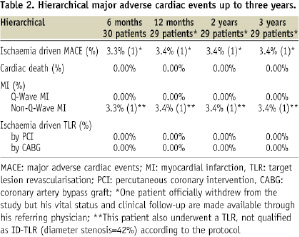
Hierarchical event rates concerning ID-MACE are presented in Table 2.
Up to three years, there was only one non-Q wave myocardial infarction related to the treatment of a non-flow-limiting stenosis (QCA DS 42%) of a BVS at 46 days post procedure. At index procedure, a BVS scaffold (3.0 x12 mm) was implanted in the distal right coronary artery, followed by post dilatation with 3.5 x 9 mm compliant balloon (Voyager) inflated at a pressure of 18 atm (nominal diameter at that level of inflation 3.84 mm). At 40 days, the patient experienced a single episode of angina at rest without electrographic evidence of ischaemia. Repeat angiography showed a 42% of diameter stenosis in the BVS by quantitative analysis (Figure 3).
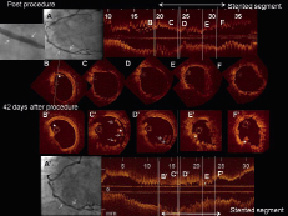
Figure 3. One patient underwent target lesion revascularisation. Post procedure, the final angiography showed a patent study stent identified with two metallic markers (Panel A, white arrow) free from residual stenosis. The OCT images were acquired post stenting but before post dilatation. Distal to the BVS, a small dissection was detected (Panel B). In the middle portion, the scaffold was well expanded with a circular shape and struts were well apposed (Panels C, D, F). At the site of the minimal luminal area (E), the lumen shape was triangular. Forty-two days after the procedure, the patient underwent repeat angiography due to recurrent angina, which revealed a moderate stenosis in the previously implanted BVS (arrow indicates metallic markers, Panel A’). The OCT cross-sections (B’-F’) were matched to the post-procedural OCT images (B-F). At the distal edge, the small dissection remained partially unhealed (asterisk, Panel B’). In the middle of the scaffold (C’), struts are detached from the vessel wall (arrow), with an irregular intraluminal defect attached to the strut (asterisk). Two strut rows (D’, arrows) are overhung and malapposed in parallel, which suggests a disruption of scaffold structure. At the previous site of minimal luminal area, the lumen was deformed into a triangular shape (E’), due to late recoil of the scaffold. Occasionally, irregular intraluminal defects attached to the vessel wall were observed with attenuation (6-7 o’clock, asterisk, F’).
An attempt to cross the stenotic lesion with the pressure wire failed: the pressure wire seemed to get entangled in the scaffold strut at the site of the stenosis. Subsequent OCT image acquisition using a Helios catheter disclosed the following: i) unhealed distal edge dissection, ii) two rows of overhung and malapposed struts, suggesting strut fracture, iii) minimal luminal area of 2.7 mm2, due to reduction of treated area by 33% in comparison with baseline measurement, iv) late acquired struts malapposition, v) irregular intraluminal defects attached to the vessel wall. Although there was no evidence of ECG ischaemia at rest, for a perceived safety reason the polymeric scaffold was covered by a drug-eluting metallic stent (non-ID TLR). After this repeat revascularisation procedure, the patient had a slight rise in cardiac enzymes (peak troponin 2.21 ng/ml, CK-MB 87.7 µg/L), which was adjudicated as a non-Q wave MI. The patient died from Hodgkin’s disease at day 888 (non-cardiac death).
Overall, there were no new MACE events between six months and three years. In the entire cohort, there was no instance of stent thrombosis according to either the protocol or ARC definitions, a retrospective analysis requested by the Food and Drug Administration at the end of 2006.13,14
Non-hierarchical event rates are shown in Table 3.
Two patients died from non-cardiac cause: one at 888 days from Hodgkin disease as described above, and the other at 706 days from a duodenal perforation. The latter patient was hospitalised on day 699 because of a haemoglobin drop due to bleeding of the upper gastrointestinal tract presumably related to intermittent use of non-steroid anti-inflammatory drugs. On day 703, the patient experienced a duodenal perforation and underwent surgery, which was not curative with persisting peritonitis. The patient refused a second intervention and died on day 706. This patient had stopped clopidogrel 364 days post procedure and was not taking aspirin since he has been on anti-vitamin K prior to his implantation.
Two patients underwent target vessel revascularisation for lesions with DS <50% prior to vasomotion test (Figure 4).
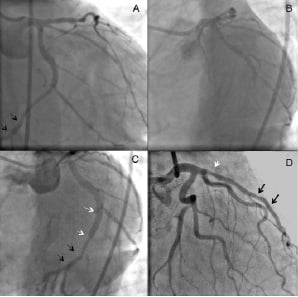
Figure 4. Two patients underwent non-ischaemic target vessel revascularisation. One patient with a BVS in the distal left circumflex artery underwent repeated angiography that revealed a patent BVS (black arrows, A: caudal angiographic view) without stenosis. The diagnostic methergine test resulted in subtotal occlusion of the middle left circumflex artery distal to the take-off of the postero-lateral branch, due to severe vasospasm (B: left oblique angiographic view). The spastic segment after relief of spasm by intracoronary injection of nitrate was treated with Taxus stent (3.5 x 20 mm, white arrows in Panel C: left oblique angiographic view). IVUS revealed that the gap between the previous BVS and the Taxus was 7 mm, therefore this procedure was adjudicated as non-target lesion, target vessel revascularisation. The other patient received a BVS in the distal LAD (black arrows, Panel D). The patient underwent repeat angiography due to recurrent angina, which demonstrated a moderate non-significant stenosis in the proximal LAD (red arrow, QCA %DS <50%). The lesion was nevertheless treated with a Taxus stent (3.5 x 12 mm).
At 106 days post procedure, one patient, who had received a BVS in the distal left circumflex artery, underwent angiography to elucidate episodes of angina in the early hours of the morning: the coronary angiogram revealed good patency of the epicardial vessels including the segment treated with the BVS. In absence of a fixed stenosis on this diagnostic angiogram, a vasomotion test was performed with ergonovin that elicited a subtotal occlusion due to severe coronary spasms in the coronary segment proximal to the BVS, which was fully relieved by intracoronary administration of nitrate. The decision was made to implant a metallic drug-eluting stent in the spastic proximal LCx lesion. Since the metallic stent was placed more than 5 mm proximal to the BVS, the event was adjudicated as a non-ischaemia driven target vessel revascularisation according to the protocol (non target lesion) (Figure 4B). The other patient had received a BVS in the distal left anterior descending (LAD) artery. On day 85, this second patient underwent a diagnostic angiography because of stable angina Class II, that disclosed a moderate, non-significant stenosis (QCA %DS <50%) in the LAD far proximal to the patent BVS. The lesion in the proximal LAD was treated with paclitaxel-eluting stent, and this procedure was adjudicated as a non-ischaemia driven target vessel revascularisation (non-target lesion) according to the protocol (Figure 4C).
The protocol mandated a minimum of six months duration of combined anti-platelet therapy but the actual duration was left to the discretion of the investigators who, in general, followed the guidelines of the ESC/ACC/AHA for anti-platelet use after metallic drug-eluting stent implantation.15,16 At one year, fifteen patients were on dual anti-platelet therapy, while one patient was kept on clopidogrel at two and at three years (Figure 5).
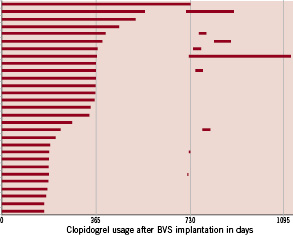
Figure 5. The bar graph demonstrates the duration of thienopyridine in individual patients. In seven patients, thienopyridine was restarted at 2-year angiography and was continued more than three days.
In patients who took clopidogrel after one year, there were no specific reasons recorded for prolonged prescription of clopidogrel.
Discussion
This short report on the 3-year clinical outcomes of the ABSORB trial demonstrates a low ischaemia-driven MACE rate of 3.4% without any stent thrombosis, early, late or very late. There is only one patient who experienced a non-ischaemia driven TLR and subsequently a procedure-related non-Q wave MI. There were no additional ID-MACE between six month and three years.
CEC adjudicated this repeat revascularisation as a non-ischaemia driven TLR according to the protocol (see the method) designed in 2005. The clinical event definition created by the academic research consortium and embraced by FDA were not available at the time of the trial design.13,14
This single failure raises the following important question: Why did this particular stent exhibit shrinkage of 33% in the stent area? – the highest stent area reduction observed at six months in the present series. Polymeric scaffolds are size-limited. The BVS studied in this trial cannot be dilated beyond 3.5 mm – as performed in this case (see above, the result section) – or it may show signs of strut fracture, potentially disrupting the mechanical integrity and radial strength of the scaffold. The OCT images obtained in in vitro experiments (Figure 6) resemble the one observed in our patient at day 42, suggesting that early “restenosis” was presumably the result of early recoil actually not prevented by the disrupted scaffold. No control OCT was performed in this patient after post dilatation during the index procedure, so that the fracture of the struts was potentially not detected at that moment.
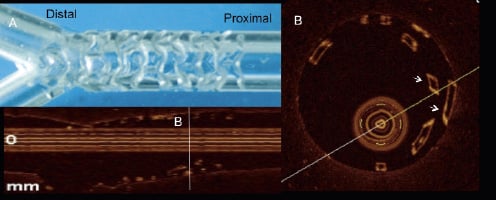
Figure 6. OCT images of strut fracture in a BVS (3.0 mm x 18 mm) implanted in a silicone phantom model. The stent was overstretched by an oversized balloon (3.5 mm in diameter, Panel A). In the proximal part of the scaffold, broken struts appeared as a double row of overhung struts malapposed to the phantom wall (arrows, Panel B).
Forty-two days later, there were intraluminal struts protruding into the vessel lumen presumably due to the possible fracture of struts (Figure 3). A single case of suspected stent fracture suggests that careful and adequate sizing of vessel diameter are of paramount importance for the correct performance of this scaffolding technique and require accurate assessment of vessel dimension by QCA prior to implantation.
This is the longest available follow-up of the first cohort of 30 patients who were treated with a fully bioresorbable everolimus-eluting scaffold. The 3-year clinical outcomes are quite encouraging: ID-MACE is as low as 3.4% without any late complications. Importantly, there is no stent thrombosis after cessation of thienopyridines (Figure 5). Since the polymeric struts are fully resorbed, there would be theoretically less risk of stent thrombosis after resorption. In the two-year OCT assessment of the BVS in the ABSORB cohort, a large number of polymeric struts were no longer recognisable. In the animals sacrificed at two years following OCT investigation (unpublished data), the polymer material in the strut voids was no longer detectable by post mortem chromatography, while the struts were still recognisable with in vivo OCT. A thienopyridine therapy shorter than two years might be therefore suitable, and the optimal duration of antiplatelet therapy should be further investigated according to either resorption or coverage of the resorbable device.
The favourable clinical results demonstrated in this study need to be further confirmed in a large study. Currently, an extension of the first-in-man study is being conducted. In the ABSORB Cohort B trial, a new version of the BVS (BVS revision 1.1) was used, with modification in strut design to enhance the mechanical properties of the scaffold and to reduce either acute or late recoil, which was suggested to be inferior to the metallic stent8,9. The BVS revision 1.1 uses the same polymers as in the original 1.0 design but with increased process controls. The strut thickness remains the same; however, the new design has in-phase zigzag hoops linked by bridges. The ABSORB Cohort B recently completed its enrolment with 101 patients in 12 sites in the European Union, Australia and New Zealand. These patients were divided into two groups, the first group (45 patients) having imaging follow-up procedures performed at 180 days and two years and the second group (56 patients) having imaging follow-up procedures performed at one year and two years. Based on the results of this second cohort of patients, the future clinical trial strategy including pivotal trials of a bioresorbable vascular scaffold (BVS) against a metallic drug-eluting stent will be developed.
Acknowledgements
We would like to thank Cecile Dorange, Karine Miquel-Hebert and Krishnankutty Sudhir for their intellectual input in reviewing the manuscript.
Appendix
Clinical Events Committee: C. Hanet, MD, PhD, Brussels, Belgium; D. McClean, MD, Christchurch, New Zealand; V. Umans, MD, PhD, Alkmaar, The Netherlands
Data Safety Monitoring Board: J. Tijssen, PhD, Amsterdam, The Netherlands; T. Lefèvre, MD, Massy, France; P. Urban, MD, Geneva, Switzerland
