Abstract
Aims: SPIRIT Women is the first interventional trial dedicated exclusively to women, focusing on symptoms at presentation, referral time to coronary intervention and the safety and performance of the XIENCE V stent.
Methods and results: SPIRIT Women is a prospective, open-label, multicentre study in which 1,573 women were enrolled at 73 sites outside the United States. The primary endpoint is the composite of all death, Academic Research Consortium (ARC) defined myocardial infarction (MI) and target vessel revascularisation (TVR) at one year. Data collected included symptoms at presentation and referral to coronary intervention. To allow comparison by gender, the latter were compared to data from male patients from the SPIRIT V study. The one- and two-year composite of all death, MI and TVR was 12% and 15%, respectively. Target lesion revascularisation (TLR) and stent thrombosis (definite and probable) rates were 2.4% and 0.59%, respectively, at one year and 3.6% and 0.73%, at two years. The total referral time for coronary intervention in women was four days longer than for men in the SPIRIT V study.
Conclusions: The XIENCE V stent is safe and effective with low TLR and stent thrombosis rates. More efforts remain to be made to increase the awareness of women and physicians of the risk for coronary artery disease (CAD).
Abbreviations and acronyms
ARC: Academic Research Consortium
CAD: coronary artery disease
EES: everolimus-eluting stent
MI: myocardial infarction
PCI: percutaneous coronary intervention
TLF: target lesion failure
TLR: target lesion revascularisation
TVR: target vessel revascularisation
SAS: single arm study
Introduction
Women generally perceive breast cancer as their leading killer. However, the main cause of death in women of all ages is in fact cardiovascular disease1, but general awareness is low. It has been reported that less than 20% of physicians are aware that annual mortality due to cardiovascular disease was higher in women than in men2. Despite the prevalence of cardiovascular disease being similar in both sexes1, it still appears to be a seriously under-recognised issue for women. Women continue to be underdiagnosed and undertreated3,4. In addition they are always under-represented in cardiovascular clinical trials, which leads to a lack of reliable evidence for proper management of cardiovascular disease in the female population3. Indeed, as the pathophysiology, clinical presentation, response to therapies, and adverse outcomes in women may differ from those in men3,5-9, caution should be exercised when extrapolating cardiovascular data for women based on clinical trials performed primarily with men10.
The XIENCE V everolimus-eluting stent (EES) has been studied extensively in the SPIRIT family of clinical trials. Recently, the single arm study, SPIRIT V, showed its safety and effectiveness in higher risk patients with multiple, complex de novo lesions11. Female representation in these studies was relatively low and ranged from 22% in the SPIRIT V11 study to 32% in the SPIRIT III12,13 study.
SPIRIT Women is the first prospective, large interventional study to exclusively focus on female patients to help address these issues. This paper reports on the clinical presentation, referral time for coronary intervention and the one- and two-year clinical outcomes with EES in female patients.
Methods
Study design
The SPIRIT Women study design has been previously published10. This single arm study (SAS) is a prospective, open label, single arm, multicentre study designed to evaluate the safety and performance of the EES in the treatment of female patients with CAD, according to its instructions for use. In the SAS 1,573 female patients, derived from the general interventional cardiology population and admitted for a percutaneous coronary intervention (PCI) procedure, were enrolled at 73 centres outside the United States. The randomised multicentre substudy comparing the EES to the CYPHER® Select™ Plus sirolimus-eluting coronary stent (Cordis, Johnson & Johnson, Warren, NJ, USA) will be reported separately.
To allow comparison by gender (for baseline demographics, symptoms at presentation and referral time analysis only), data from male patients from the prospective, open label, multicentre SAS of the SPIRIT V study was used. In the SAS, 2,663 patients, of whom 78% were men (n=2,072) were enrolled in 93 sites outside the United States. The design of this study has been described in detail elsewhere11. The SPIRIT V and SPIRIT Women trials collected the same data concerning symptoms and the pathway to referral.
PATIENTS
Included in the trial were patients aged >18 years, with evidence of myocardial ischaemia, stable or unstable angina or silent ischaemia. Patients had to be acceptable candidates for coronary artery bypass graft surgery and needed to agree to undergo all protocol-required follow-up examinations. For patients of child-bearing potential, a negative pregnancy test within seven days before treatment was required. A maximum of four planned study stents was allowed for treatment of de novo lesions with reference vessel diameter between 2.25 and 4.0 mm and lesion length ≤28 mm (by visual estimation). Key exclusion criteria were previous participation in another device or drug study or completion of the follow-up phase of another study within 30 days prior to enrolment, and previous stent implant of either a bare metal stent or a drug-eluting stent within the target vessel.
All patients were required to provide written informed consent prior to study inclusion, as approved by the appropriate medical ethics committee of the respective clinical site.
PROCEDURAL INFORMATION
Following confirmation of the angiographic inclusion criteria and before implantation of the first stent, patients were registered via an interactive voice response system (ICON Clinical Research, Eastleigh, UK). All registered patients were considered enrolled in the study and were to remain in the study until completion of the required follow-up period.
Details of the EES have been published previously14. The stent consists of a cobalt chromium alloy platform with a drug-eluting coating, which is composed of two polymers and the antiproliferative drug everolimus. It was available in diameters of 2.25, 2.5, 2.75, 3.0, 3.5, 4.0 mm with lengths of 8, 12, 15, 18, 23, 28 mm. The treatment strategy was determined by the investigator. A target lesion was defined as any lesion to be treated at the time of the index procedure. All target lesions were to be treated with the EES. If any staged procedures were planned at baseline, or in the event of bailout and additional stent requirement, an EES of appropriate length was to be used.
Periprocedural pharmacotherapy was administrated according to standard hospital practice. Either unfractionated heparin or bivalirudin was allowed for procedural anticoagulation. The use of glycoprotein IIb/IIIa inhibitors was left at the discretion of the investigator. Regarding antiplatelet medication, the pre-procedure loading dose was ≥300 mg for clopidogrel and ≥75 mg for aspirin. Post-procedure, a daily dose of 75 mg of clopidogrel (minimum six months) and ≥75 mg of aspirin (indefinitely) were recommended. Clinical follow-up was scheduled at 30 days, 240 days, one and two years.
SOURCE DOCUMENT VERIFICATION
At each site, 20% of patient data were monitored against source documents and 100% source verification was performed on patient informed consent forms and reported adverse events. All endpoint-related events reported were adjudicated by an independent clinical events committee (Appendix 1).
PATIENT HISTORY
Patient history was recorded and included demographics and symptoms at presentation. Information on the referral pathway to cardiac intervention was obtained by identifying all visits (i.e., cardiologist, emergency room, catheterisation laboratory, general practitioner) that had occurred since the first symptom leading to the intervention. Patients were asked to specify the duration between the first symptom and the referral points mentioned above. Patients were also questioned regarding their smoking history, menopausal status, hysterectomy and oophorectomy status, weak oestrogen use, past and present use of hormonal contraceptives/hormone replacement therapy, educational/professional/social background and chronic concomitant medications.
STUDY ENDPOINTS
The primary endpoint was the adjudicated composite rate of all death (Academic Research Consortium criteria defined15), MI and TVR at one year. Secondary endpoints included, but were not limited to, acute success (clinical device and procedure) and stent thrombosis rates.
DEATH
All deaths were considered cardiac unless an unequivocal non-cardiac cause could be established. Specifically, any unexpected death, even in patients with coexisting, potentially fatal non-cardiac disease (e.g., cancer, infection), were classified as cardiac.
CARDIAC DEATH
Any death due to immediate cardiac cause (e.g., MI, low-output failure, fatal arrhythmia). Unwitnessed death and death of unknown cause were classified as cardiac death. This included all procedure-related deaths including those related to concomitant treatment.
MI
MI classification and criteria for diagnosis were defined according to the ARC as follows: for non-procedural/spontaneous MI, troponin or creatine kinase muscle and brain (CK-MB) levels had to be >2 times the upper limit of normal; for peri-PCI, troponin or CK-MB levels had to be ≥3 times the upper limit of normal; for peri-coronary artery bypass graft (CABG), troponin or CK-MB levels had to be ≥5 times the upper limit of normal. The periprocedural period included the first 48 hours and 72 hours after PCI and CABG, respectively. All late events that were not associated with a revascularisation procedure were considered spontaneous. One blood sample was taken from each patient within the post-procedure hospitalisation period for the analysis of CK-MB or troponin levels.
TARGET LESION REVASCULARISATION
TLR is defined as any repeat percutaneous intervention of the target lesion or bypass surgery of the target vessel performed for restenosis or other complications of the target lesion. The target lesion is defined as the treated segment from 5 mm proximal and 5 mm distal to the stent.
TARGET VESSEL REVASCULARISATION
TVR is defined as any repeat percutaneous intervention or surgical bypass of any segment of the target vessel. The latter is defined as the entire major coronary vessel proximal and distal to the target lesion including upstream and downstream branches and the target lesion itself.
TARGET LESION FAILURE (TLF)
TLF is defined as the composite of cardiac death, target vessel MI, and ischaemia-driven TLR (PCI or CABG).
STENT THROMBOSIS
Stent thrombosis was categorised as acute (<1 day), subacute (1-30 days) and late (>30 days) and was defined according to the ARC guidelines as follows: definite - acute coronary syndrome and angiographic or pathologic confirmation of stent thrombosis; probable - unexplained death ≤30 days or target vessel myocardial infarction (TV-MI) without angiographic information; and possible - unexplained death >30 days after stent placement.
CLINICAL DEVICE SUCCESS
Clinical device success was defined as being the delivery and deployment of the study stent (in an overlapping stent setting, the delivery and deployment of the first and following study stent) at the intended target lesion and withdrawal of the stent delivery system with attainment of final residual stenosis of less than 50% of the target lesion by quantitative coronary angiography (QCA) (by visual estimation if QCA unavailable), without use of a device outside the assigned treatment strategy. Bailout patients were included in this clinical device success category only if the above criteria were met.
CLINICAL PROCEDURE SUCCESS
Clinical procedure success was defined as being delivery and deployment of the study stent or stents at the intended target lesion and successful withdrawal of the stent delivery system with attainment of final residual stenosis of less than 50% of the target lesion by QCA (by visual estimation if QCA unavailable) and/or using any adjunctive device without the occurrence of cardiac death, MI not clearly attributed to a non-target vessel and/or CI-TLR during the hospital stay with a maximum of first seven days post-index procedure. In a multiple lesions setting each lesion had to meet clinical procedure success criteria.
Statistical analysis
All analyses were performed on the intent-to-treat population. For binary variables, percentages were calculated. For continuous variables, means and standard deviations, median and interquartile ranges are presented. For the composite of death, MI and TVR, a survival curve was constructed using Kaplan-Meier estimates.
Comparisons between groups were done using the Wilcoxon rank sum test for continuous variables and the Fisher’s exact test for dichotomous variables.
The impact of several possible predictor variables on time since first symptoms were assessed comparing the different subgroups. Outliers were not considered in these analyses. The data presented are based on the SPIRIT Women study data only. In addition, the assessment of gender as a predictor was performed on the pooled data from the SPIRIT Women study and the male patients from the SPIRIT V study.
Results
A total of 1,573 patients comprised the intent-to-treat population (ITT). At 240 days, four patients had withdrawn informed consent, five had been lost to follow-up and one patient did not require follow-up, leaving a population of 1,563 patients. At one year, three patients had withdrawn informed consent and three patients had been lost to follow-up, leaving a population of 1,557 patients (99%). At two years, two patients had withdrawn informed consent, one patient refused the follow-up and 15 patients had been lost to follow-up, leaving a population of 1,539 patients (98%) (Figure 1). As defined by the protocol, all results are presented for the ITT population (which included all patients who had signed the informed consent, had been registered via the interactive voice response system and who had at least one EES implanted, a non-EES implanted following an attempt to implant an EES, or no stent implanted following an attempt to implant and EES).

Figure 1. Clinical study population. In the SAS of the SPIRIT Women study, 1,573 female patients were recruited in 73 centres outside the United States and comprised the intent-to-treat population. The 1- and 2-year clinical follow-up was completed by 1,557 patients (99%) and 1,539 (98%) patients, respectively.
PATIENT CHARACTERISTICS, SYMPTOMS AT PRESENTATION AND REFERRAL PATHWAY FOR CORONARY INTERVENTION (THE SPIRIT WOMEN TRIAL VS. MALE PATIENTS FROM THE SPIRIT V TRIAL)
At the time of presentation, women were on average five years older than men (67 years vs. 62 years) (Table 1). In addition, women were at increased risk for CAD as evidenced by the higher rate of hypertension treated with medication (78% vs. 62%), hypercholesterolaemia treated with medication (64% vs. 58%) and diabetes (34% vs. 28%). Of significance, 26% of women had experienced a previous MI vs. 33% of men with 45% vs. 14% of these within the two months prior to the index procedure. Insulin-dependent diabetes mellitus (IDDM) was observed in 11% of female patients compared to 6% in men. General obesity, defined as a body mass index (BMI) >30 kg/m², was observed at an equally high rate in women (27%) and men (24%) with a mean BMI of 28 kg/m² in both sexes. Central obesity, defined as a waist circumference of >88 cm in women and >102 cm in men, however, is considered a more important CAD risk factor. Seventy-one percent of the female patients were centrally obese (mean waist circumference: 95±14 cm) compared to 39% of men (mean waist circumference: 99±11 cm).
When compared to men, more women reported atypical angina (9% vs. 6%) or no chest pain at all (17% vs. 13%) (Figure 2). To gain more information on the referral pathway for coronary intervention, patients were asked to identify the points of referral (i.e., general practitioner, cardiologist, emergency room, catheterisation laboratory) since the first cardiac symptom experienced and to specify the duration between this first symptom and each referral point. The median duration between the onset of symptoms and the first referral point was four days (Q1=0.17, Q3=20) in males compared to five days (Q1=1, Q3=15) in females (p=0.02). Of interest, this difference was more marked when considering the duration between the first and last referral point: median of 0 days (Q1=0, Q3=5) for male patients vs. one day (Q1=0, Q3=9.9) for female patients (p<0.0001). The median number of days between first symptoms to last referral point was seven (Q1=3, Q3=28) and 11 (Q1=3, Q3=31) in males and females, respectively (p=0.0003).
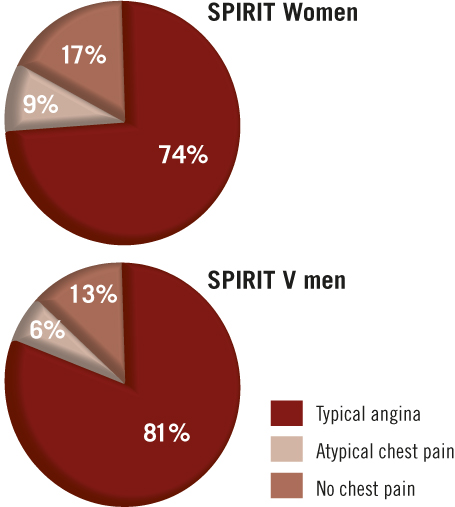
Figure 2. Presenting symptoms in women and men. The presence of typical angina, atypical chest pain or no chest pain in women and men were recorded at time of presentation.
Subgroup analyses were performed to identify variables affecting the referral time (Table 4). Smoking, unstable angina (Braunwald class III), male gender, MI within two months, having the emergency room as first referral point and typical symptoms resulted in shorter referral times, whereas hypertension, hypercholesterolaemia, stable angina (Canadian Cardiovascular Society [CCS] classification class III or IV) and seeing either the cardiologist or the general practitioner as the first referral point resulted in longer referral times. Variables such as age, prior cardiac interventions, prior MI, presence of diabetes and employment status had no effect on referral time.
ONE- AND TWO-YEAR CLINICAL OUTCOMES
Baseline patient characteristics and risk factors are presented in Table 1. The clinical procedure success (per patient) was 92% and clinical device success (per target lesion) was 99% (Table 2). The majority of treated lesions were located in the left anterior descending (LAD) artery (48%) with 1% of lesions located in the left main coronary artery. Target lesions were ≥20 mm in 29% of the patients and the mean lesion length was 15 mm. The mean reference vessel diameter (RVD) was 2.9 mm with 47% of the patients having a RVD ≤2.75 mm. An average of 1.6±1.0 stents were implanted per patient. Thrombus and moderate to severe calcifications were reported in 6% and 30% of the lesions, respectively. Seventy-two percent of lesions were American College of Cardiology (ACC) classification B2 or C.
The primary composite endpoint of all death, ARC-defined MI and clinically indicated TVR at one year was 12.1% (Table 3, Figure 3) and 14.8% at two years. The rate of this composite endpoint was primarily driven by the high hierarchical MI rate of 9.0% and 9.3% at one year and two years, respectively. Of 154 patients experiencing MI events, 112 were peri-index procedural. Target lesion failure (TLF), defined as composite of cardiac death, MI related to the target vessel and TLR rate was 10.7% at one year and 12.5% at two years. This rate was again primarily driven by the hierarchical rate of MI not clearly attributed to a non-target vessel (8.6% and 8.8% hierarchically at one and two years, respectively). The non-hierarchical ARC-defined periprocedural MI rate was 7.1% (112 out of 1,573 patients). The overall TLR rate was 2.4% at one year and 3.6% at two years and the cumulative stent thrombosis (definite and probable) rates were respectively 0.59% and 0.73% (Figure 4). At one-year follow-up, 90% of the patients were still on clopidogrel and 96% of the patients were taking aspirin. At two-year follow-up, only 62% of the patients remained on clopidogrel, and aspirin medication was ongoing for 94% of the patients.
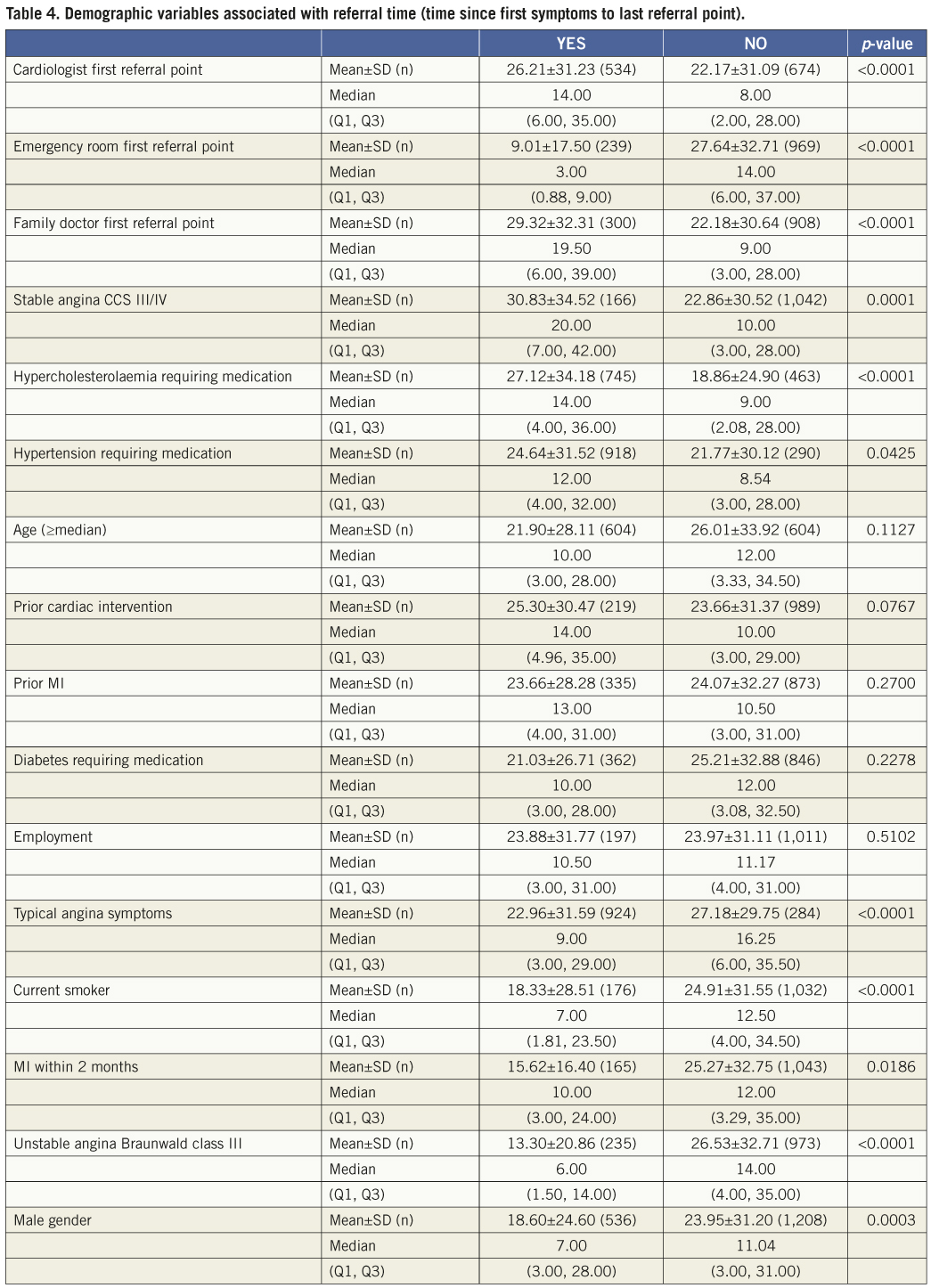
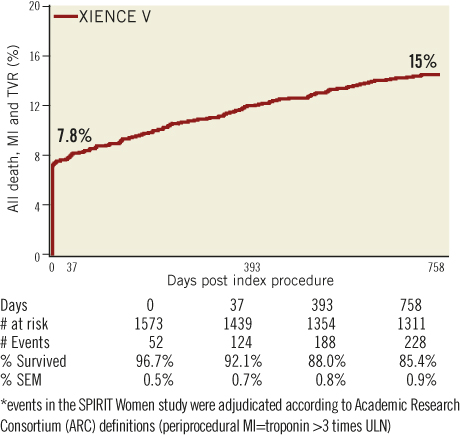
Figure 3. ARC-defined primary endpoint. Kaplan-Meier estimates were used to construct a survival curve for the composite of death, MI and TVR.
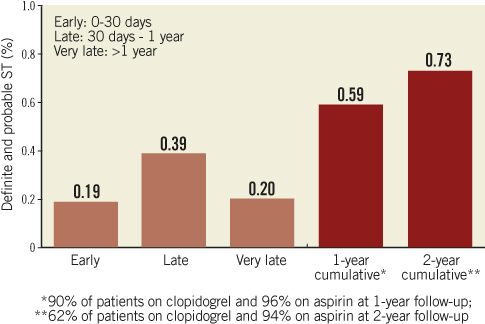
Figure 4. Stent thrombosis rates. Stent thrombosis rates, categorised as acute (<1 day), subacute (1-30 days), late (>30 days) and very late (>1 year), were defined according to the ARC guidelines as described in the methods section.
Discussion
Whether or not female gender is an independent predictor for worse outcome after PCI is still a subject of debate. Some studies confirm this view16-18, whereas others have shown that the poorer outcomes in women are due to the presence of more clinical risk factors and comorbidities19-21. A number of these studies, however, relate to balloon angioplasty in the pre-stent era, whereas others were done retrospectively and included only a small number of female patients. To date, no prospective study has specifically focused on the evaluation of drug-eluting stents in female patients.
In line with previous reports3,22-24, the women enrolled in the study were on average five years older at presentation than men and overall had more CAD risk factors. More women than men had comorbidities including hypertension, hypercholesterolaemia and diabetes and almost twice as many women as men were centrally obese.
Despite recent attempts to increase the general awareness regarding the risk of women developing CAD, data from the SPIRIT Women trial demonstrate that the total referral time (i.e., time between first symptoms and last referral point prior to revascularisation) in women is on average four days longer than in the men in the SPIRIT V trial.
In the past, the fact that women often experience more vague and atypical symptoms, including mid-back pain, nausea, fatigue, dyspnoea, palpitations and indigestion22,25-28, has been suggested to contribute to the under-diagnosis of CAD and longer referral times for coronary intervention. Indeed, when experiencing atypical symptoms, the link with CAD might be less clear for the patient and/or the physician, which in turn could lead to a delay in seeking specialised help29 and under-diagnosis. Our data, however, are more in line with other studies that report less clear-cut sex-based differences in symptoms at presentation30,31. The majority of women and men in our study presented with typical angina symptoms. A trend was seen towards more women presenting with atypical chest pain or no chest pain at all; however, these differences were small in amplitude. In contrast to previously published data, mid-back pain and pain in the arm or shoulder were reported more frequently by male patients in our study, whereas dyspnoea was reported at an equal rate in both sexes. The women in our study generally also reported a lower number of symptoms, which is not in line with a previous report by Milner and colleagues22. A possible explanation for the conflicting data on this topic may be that the patient population in some of the studies was relatively small22 and some studies only included patients with acute coronary ischaemia and acute MI25,27,28,31. Dedicated studies, including both male and female patients, could generate more conclusive information on gender-related differences in the referral pathway. There is an important need for these kinds of studies, as subgroup analyses on the SPIRIT Women trial data identified, for example, that women who presented with typical angina symptoms had a significantly shorter referral time than those who experienced atypical angina symptoms. Importantly, it also showed that women presenting with hypertension and hypercholesterolaemia had significantly longer referral times whereas diabetic women had a similar referral time compared to their non-diabetic counterparts. Since hypercholesterolaemia, hypertension and diabetes are known risk factors for developing CAD, shorter referral times for these patients may be expected.
The SPIRIT Women trial is the first prospective analysis of the safety and effectiveness of the XIENCE V stent in a female population. In addition the patients had more complex lesion types than in previous trials, and were selected under less restrictive criteria and thus more closely resembled the real world situation. The majority of women presented with known CAD risk factors, including hypertension, hypercholesterolaemia and diabetes and had a relatively high lesion risk profile. Nevertheless, the one-year clinically-indicated TLR and stent thrombosis rates were low and are consistent with the favourable safety of the XIENCE V stent that was shown in previous all-comer studies. This low rate was maintained at two-year clinical follow-up. Importantly, as there was no angiographic follow-up, all revascularisations were considered ischaemia-driven. The rate of the primary composite endpoint of all death, MI and TVR was higher (12.1%) at one year than that reported in previous studies evaluating the XIENCE V stent. This may be explained by the use of the ARC definition in the SPIRIT Women study for adjudicating MI events (baseline troponin
Previously, a gender-based performance evaluation of the XIENCE V stent was performed on the SPIRIT III trial data18. At one year, women had significantly higher major adverse cardiac events (MACE), TVF, TVR and TLR rates compared with men. Women treated with a XIENCE V stent had better angiographic and clinical outcomes than women treated with a TAXUS® paclitaxel-eluting stent (Boston Scientific, Natick, MA, USA). However, the SPIRIT III study was underpowered to definitely demonstrate a reduction in clinical revascularisation in women by the XIENCE V stent and an insufficient number of women were studied (32%; 314/1,002). In addition, patients in the SPIRIT III trial were selected according to relatively strict inclusion/exclusion criteria, excluding patients with a higher risk profile (diffuse disease, multiple and complex lesions). In a pooled gender-based analysis on the SPIRIT II and III trial data, the XIENCE V compared to the TAXUS stent resulted in reduced angiographic late loss (LL), fewer MACE and TVF events in women, and reduced angiographic LL and diameter stenosis percent (%DS) and fewer MACE events in men at two years33. Conversely, an analysis of approximately 10,000 patients treated with the TAXUS stent, including more than 3,000 women, demonstrated that despite their higher-risk profile, women have comparable benefits to men from percutaneous coronary intervention with a TAXUS paclitaxel-eluting stent except for a slightly higher revascularisation rate in the high-risk cohort34.
In the SPIRIT Women trial, 1,573 “all-comer” female patients were evaluated, which represents a large sample size. The data from the SPIRIT Women trial show that, despite the worse cardiovascular profile of female patients and the high complexity of lesions treated, the XIENCE V stent can be used safely and effectively in women, with an extremely low one-year rate of stent thrombosis (0.59%) and TVR (3%), which were maintained through two years (0.73% and 4.8%, respectively).
Strengths and limitations of the study
This study was limited by the lack of a control arm for direct comparison. The SPIRIT Women trial was designed to look at specific aspects of women’s health in relation to CAD. Inherently, it was not designed to look at gender differences in the referral pathway, referral time and symptoms at presentation. In this manuscript, comparison by gender was made against a “concurrent control”, i.e., data from male patients enrolled in the SPIRIT V single arm study. The data related to gender differences described in this manuscript should be considered hypothesis-generating and dedicated studies are required to address these gender differences in more detail and to provide more conclusive answers. In the SPIRIT V trial, the questionnaire used in the SPIRIT Women study to gather information on the referral pathway, symptoms at presentation and diagnostic tests, was added retrospectively to the start of patient enrolment. Therefore, of the 2,077 male patients enrolled in the SPIRIT V single arm study, 694 prospectively-completed questionnaires were obtained.
Countries and sites were not identical in the SPIRIT V single arm study and the SPIRIT Women study. Therefore, geographical differences in disease management, health care access and referral for diagnostic testing, which most likely exist33, were not considered in the current analysis.
Although consecutive eligible patients were to be included in the study, it cannot be excluded that a selection bias may have occurred at the time of enrolment.
The strengths of this study include the large sample size of female patients (n=1,573) and the rigorous attention to event reporting and follow-up of patients.
Conclusions
Although females have a higher risk profile for developing CAD, their referral time is longer than that for the male patients in the SPIRIT V trial. Surprisingly, women that presented with well-known CAD risk factors including hypercholesterolaemia, hypertension or diabetes, did not have shorter referral times than those women presenting without these comorbidities. This demonstrates the need for continuing efforts to increase the awareness among patients, nurses and physicians with regard to cardiovascular disease in women as earlier diagnosis could improve the prognosis in female patients. The data from the SPIRIT Women study show the safety and effectiveness of the XIENCE V stent in female patients through two-year follow-up.
Appendix
Principal investigator: Marie-Claude Morice
Co-principal investigator: Stephan Windecker
Steering committee: Maria Grazia, Modena, Italy; Ghada Mikhail, United Kingdom; Fina Mauri i Ferré, Spain; Ruth Strasser, Germany; Liliana Grinfeld, Argentina
Clinical events committee (CEC): C. Hanet, Brussels, Belgium; V. Umans, Alkmaar, The Netherlands; K. Koch, Amsterdam, The Netherlands; D. McClean, Christchurch, New Zealand; E. McFadden, Cork, Ireland; A. Hoye, Hull, United Kingdom; P. Agostoni, Utrecht, The Netherlands
Data safety monitoring board (DSMB): J. Tijssen, Amsterdam, The Netherlands; W. Wijns, Aalst, Belgium; P. Urban, Geneva, Switzerland; K. Fox, Edinburgh, Scotland
The following investigators and institutions participated in the SPIRIT Women study (numbers of patients enrolled):
Mainar V, Hospital General de Alicante, Spain (70); Amoroso G, OLVG, The Netherlands (70); Kumar S A, CARE Hospital, India (68); Suttorp M J, Stichting Sint Antonius Ziekenhuis, The Netherlands (51); Morice M-C, Institut Cardiovasculaire Paris-Sud and Institut Hospitalier Jacques Cartier Service d’Angiographie, France (49); Pasotti E, Cardiocentro Ticino, Switzerland (48); Stammen F, Heilig Hart Ziekenhuis Roeselare, Belgium (46); Abizaid A, Instituto de Cardiologia Dante Pazzanese, Unidade II Recepcao de Angioplastia, Brazil (46); Erglis A, Latvian Center of Cardiology, P. Stradina University Hospital, Latvia (41); Mauri J, Hospital Trias y Pujol, Spain (40); Glogar D and Lang I, Allgemeines Krankenhaus der Stadt Wien, Austria (38); Horvath I G, University of Pécs/Medical School/Heart Institute, Hungary (37); Merkely B, Semmelweis University, Cardiovascular Center, Hungary (37); Hernandez R, Hospital Clinico San Carlos, Spain (35); Noble J, Hôpitaux Universitaires de Genève, Switzerland (35); Richardt G, Segebergerkliniken, Germany (31); Blomerus P, Unitas Hospital, South Africa (30); Pansieri M, Hôpital Henri Duffaut, France (29); Lee M, Queen Elisabeth Hospital, Hong Kong (29); Goel P K, Sanjay Gandhi Post Graduate Institute of Medical Sciences, India (28); Zaman A, Freeman Hospital, United Kingdom (28); Lemos P, INCOR, Brazil (24); Strasser R, Dresden HZ Medizinische Klinik II - Kardiologie, Germany (24); Darremont O, Clinique Saint Augustin, France (23); Wiemer M, Bad Oeynhausen, Germany (22); Cottin Y, Hôpital du Bocage CHU, France (21); Chen J L, Fu Wai Hospital, China (21); Bartorelli A, Centro Cardiologico Monzino, Italy (21); den Heijer P, Amphia Hospital, The Netherlands (21); Brachmann J, Klinikum Coburg GmbH, Germany (20); Buszman P, Polsko Amerykanskie Kliniki Serca American Heart Clinics, Poland (20); Torres J, Clinica Santa Sofia, Venezuela (20); Grinfeld L, Hospital Italiano de Buenos Aires, Argentina (19); Modena M G, Ospedale di Modena, Italy (19); Lee S, Queen Mary Hospital, Hong Kong (18); Belardi J, Instituto Cardiovascular de Buenos Aires, Argentina (17); Chen YD, China People Liberation Army (PLA) General Hospital, China (17); Gilard M, Hôpital de la Cavale Blanche, France (17); Molstad P, Feiringklinikken AS, Norway (17); Chandra P, Max Devki Devi Heart and Vascular Institute, India (16); Wan Ahmad WA, University Malay Medical Center, Malaysia (16); Zeymer U, Klinikum Ludwigshafen, Germany (16); Rath PC, Apollo Hospital, India (15); Yue C-S, United Christian Hospital, Hong Kong (15); Cebollero IC, Hospital Miguel Servet de Zaragoza, Spain (15); Lebreton H, Centre Cardio-Pneumologique Hôpital Pontchaillou, France (14); Inglese L, Policlinico San Donato, Italy (14); Eltchaninoff H, Hôpital Charles Nicolle, France (13); Fischer D, Medizinische Hochschule Hannover, Germany (13); Jung W, Kliniken Villingen, Germany (12); Marino MA, Hospital Madre Teresa, Brazil (12); Ferreira R, Hospitalar Santa Marta, Portugal (12); Chen J, Guangdong Provincial People’s Hospital, China (11); van der Merwe N, Bloemfontein Medi Clinic, South Africa (11); Mikhail G, St Mary’s Hospital, United Kingdom (11); Chow S L, Tuen Mun Hospital, Hong Kong (10); Manari A, Azienda Ospedaliera Santa Maria Nuova, Italy (10); Schoors D, Universitair Ziekenhuis Brussel, Belgium (9); Kaehler J, UKE Hamburg, Germany (9); Genth-Zotz S, Uni Mainz, Germany (9); Silva JC, Hospital São João, Portugal (9); Garcia EF, Hospital Gregorio Marañón, Spain (8); Ludman P, Queen Elisabeth Hospital, United Kingdom (8); Rozenman Y, Wolfson Medical Center Heart Institute, Israel (7); Van Gaal B, The Northern Hospital, Australia (5); Leclercq F, Hôpital Arnaud de Villeneuve CHU, France (5); Nordrehaug JE and Tuseth V, Haukeland University Hospital, Norway (5); Hauptmann K E, Krankenhaus der Barmherzigen Brüder, Germany (4); Lalmand J, CHU Charleroi, Belgium (4); Witkowski A, Institute of Cardiology, Poland (3); Desaga M, Kreisklinik Dachau, Germany (2); Juergens C, Liverpool Hospital, Australia (1); Qiu J, Guangzhou Army General Hospital, China (1)
Funding
This study was funded by Abbott Cardiovascular Systems, Inc., a subsidiary of Abbott Vascular, Santa Clara, CA, USA.
Conflict of interest statement
F. Mauri has received research grants from Abbott Vascular; K. Sudhir, M. Stuteville, P. Papeleu, D. Li, and D. Rutledge are employees of Abbott Vascular. All other authors have no conflict of interest to declare.
