Abstract
Recently, catheter-based renal denervation (RDN) has become available. In order to understand better the possible role of RDN as a treatment modality, we first discuss the anatomy and function of the renal nerves in this brief review. Secondly, we address the question - what is the clinical evidence for the involvement of the kidneys and renal nerves in the pathogenesis of sympathetic hyperactivity. Finally, we will discuss how this sympathetic hyperactivity can be reduced, specifically addressing the possible role of RDN.
Introduction
Hypertension is an important predictor of cardiovascular mortality. Traditionally, the treatment regimen includes adjusting lifestyle (for instance, weight reduction and excess sodium intake) and medication. However, a substantial proportion of patients are not adequately controlled in this way and therefore remain at increased cardiovascular risk. Recent technological developments have made it possible to interfere with the function of the renal nerves. First results suggest that this approach could be a useful addition to presently accepted treatment regimens. Therefore, it seems appropriate to review briefly: 1) the anatomy and function of the renal nerves, 2) how to assess sympathetic activity, 3) the question whether there is clinical evidence for involvement of the kidneys and renal nerves in the pathogenesis of sympathetic hyperactivity, and 4) how this sympathetic hyperactivity can be reduced, specifically addressing the possible role of renal denervation (RDN).
The anatomy and function of the renal nerves
Sympathetic outflow towards the body is generated in the central nervous system. This is called efferent nerve activity. The function of multiple organs, including the heart, vasculature and the kidneys, is influenced by this efferent sympathetic nerve activity. There is no such thing as a “general” sympathetic outflow; sympathetic outflow to the various organs may vary greatly in that it is increased to one organ whereas it is low or absent towards another organ. The degree of efferent activity is modulated by afferent nerve signals. These are nerves coming from the various organs towards the central nervous system.
The renal nerves are located in the adventitia around the renal artery1. The kidneys are both a recipient of efferent sympathetic signals as well as a generator of renal afferent sympathetic activity. There is no evidence for parasympathetic innervation of the kidneys2. Renal nerve activity cannot be measured directly, at least not in humans. The downstream effects can be quantified and are discussed below.
FUNCTION OF EFFERENT RENAL NERVES
There is extensive experimental evidence that stimulation of the renal efferent nerves results in a cascade of actions in the kidneys: as a result of renal vasoconstriction, renal blood flow and glomerular filtration rate decrease. Moreover, sodium reabsorption increases and angiotensin II (Ang II) is produced. Ang II directly causes vasoconstriction, has trophic effects and plays an important role in the water and sodium reabsorption in the proximal tubule3. All these effects are either directly or indirectly involved in the pathogenesis of hypertension. However, it is still unclear whether increased efferent nerve activity as primary event/abnormality exists.
FUNCTION OF AFFERENT RENAL NERVES
There is convincing evidence that afferent renal nerves do exist. More specifically, they might be very important in various disease conditions.
First, we will discuss experimental evidence that supports the existence of sympathetic nerves in the kidneys. The first study that showed the importance of the renal nerves in inducing sympathetic overactivity was the study of Kottke et al, which showed that chronic stimulation of the renal artery nerves in dogs caused sustained hypertension4.
Kidney ischaemia is the central mechanism of stimulation of the renin angiotensin system (RAS) and of high sympathetic nerve activity. Even a minute lesion by injection of phenol into one kidney caused increased central sympathetic activity and hypertension5. Vice versa, RDN or unilateral nephrectomy decreased the risk of developing hypertension6. The role of sympathetic overactivity was illustrated in a model where long-term low-dose Ang II infusion in rats produced a gradual increase in blood pressure, while RDN partially prevented hypertension7. Moreover, in renal transplantation models Ang II receptors appeared to be crucial in the pathogenesis of Ang II mediated hypertension and organ damage8. Whether the effect of Ang II on sympathetic nerve activity is direct or mediated through kidney injury is unclear. Ang II acts on different levels, i.e., the kidneys, the central nervous system and peripheral sites to provoke noradrenaline release from sympathetic nerve terminals. Kidney injury, generally characterised by increased RAS activity, can increase sympathetic nerve activity and causes hypertension as well as organ damage. Increased sympathetic activity enhances the activity of the RAS, suggesting reciprocal potentiation of the two systems. For the most part, destruction of the renal nerves prevents these pathophysiologic mechanisms; therefore, renal innervation must play an important role.
Methods to quantify sympathetic activity in humans
How can we assess sympathetic activity in humans? Two methods of quantifying sympathetic activity have greatly increased our knowledge over the past 20 years.
NORADRENALINE SPILLOVER
Noradrenaline (NA) synthesis takes place in neural tissue from the amino acid tyrosine and is released from the nerve to stimulate pre- and post-synaptic adrenoreceptors. When sympathetic nerves are stimulated, more NA is released. NA can easily be measured in plasma obtained by peripheral venous sampling. However, in that case it should be considered a poor indicator of sympathetic activity, because it actually represents the end result of local release and removal. In certain experimental settings, it can be a very useful investigational tool. For instance, NA measured in renal venous blood flow was reduced by more than 90%, when renal nerves were surgically removed2. Measurement of NA and its metabolites in the urine can also be done easily and in large patient groups. It represents the end result of filtration, local production and removal. Measurement of NA, using radiotracer technology, has been used to quantify the regional sympathetic activity. This method combines intravenous infusion of tritiated NA with regional sampling. For instance, it was recognised by this technique that in early hypertension renal NA spillover was significantly elevated as compared to controls, suggesting renal sympathetic overactivity9. Further, NA spillover increased when renal afferent nerves were stimulated2. This method has been mainly applied by Murray Esler and his co-workers, who have produced a large number of important papers over more than two decades. Recently, they showed that RDN reduced renal spillover by 42%10. Some nice reviews dealing with this subject have been published11,12.
MUSCLE SYMPATHETIC NERVE ACTIVITY (MSNA)
In the 1970s, MSNA was introduced by Gunnar Wallin and co-workers. This method assesses real nerve activity, and quantifies the centrally originated postganglionic efferent sympathetic nerve activity towards the resistance vasculature13,14. This microneurography is done with a tungsten electrode, inserted into the autonomic part of a peripheral nerve, most often the peroneal nerve, together with a reference microelectrode within 2-3 cm (multi-unit MSNA). Sympathetic activity is recorded as visual and acoustic identification of bursts (multifibre discharge)15. Multiple studies showed that the signal has very high within-subject reproducibility. MSNA increases with age. It is increased in some but not all patients with essential hypertension. It is already increased in patients with early chronic kidney disease (CKD) and even more pronounced in patients with advanced kidney failure. Further, it is increased in renovascular hypertension and obesity-related hypertension16-20. Finally, there is also much evidence that heart failure patients often have increased MSNA20. The downside is that assessment of MSNA is invasive, time-consuming and it requires an investigator who is very experienced with the technique. Therefore, it is not suitable for routine use. As it has a high intraindividual reproducibility, MSNA is especially suitable to quantify the effect of an intervention within subjects21.
Clinical evidence for the role of the renal nerves in sympathetic activity
Can we extrapolate from these experimental data to humans? Yes, there is also rather good evidence that these mechanisms, mentioned earlier, also work in humans. As shown in Figure 1 various downstream effects can be expected. Converse and co-workers were the first to show that MSNA is increased in CKD patients; the method they use captures sympathetic activity originating in the central nervous system acting downstream on resistance vessels22. Of particular importance is the fact that in bilaterally nephrectomised patients MSNA was comparable to controls22,23. Renin and Ang II became undetectable after bilateral nephrectomy24. These and other studies present definite proof that diseased kidneys are of central importance in the pathogenesis of sympathetic overactivity. The interaction between the RAS and the sympathetic system is illustrated by the observation that there is parallel activation of the renin and the sympathetic systems in CKD patients20. The activity of both systems shows parallel changes in fluid status19. Moreover, intravenous infusion of Ang II stimulates MSNA in humans. These data support the hypothesis of a cause and effect relation (or a common origin)25. Even in the early stages of kidney failure MSNA levels are increased and become more pronounced when eGFR is reduced26. Sympathetic overactivity seems to be an early phenomenon in the clinical course of kidney failure. This is also observed in congestive heart failure27.
As we discussed earlier, the data taken together seem to indicate that there are (at least) two types of sympathetic activity of central origin acting on resistance vessels: 1) on the one hand “baseline” activity under control of the central nervous system and the baroreceptor, which is seen both in healthy persons and even in bilaterally nephrectomised patients; 2) on the other hand sympathetic “overactivity” as a result of stimuli originating in the diseased kidneys28. The latter closely correlates with the activation of the RAS28. What triggers the renal signal? Presumably ischaemia. Even small lesions which do not affect kidney function can lead to this chain of events5. It is therefore likely that this sequence of events, as schematically shown in Figure 1, is operational in many disease conditions, including hypertension, chronic kidney disease, heart failure and obesity.
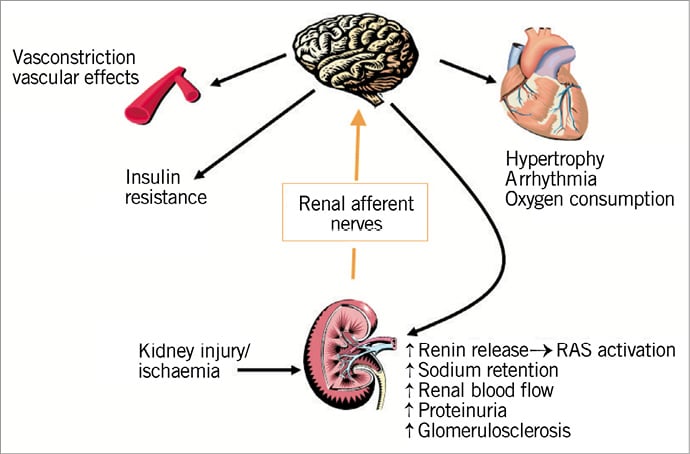
Figure 1. Schematic representation of the kidney involvement in the pathogenesis of sympathetic hyperactivity.
Taking all the data together, there is very convincing evidence that kidney injury and/or kidney ischaemia is the key feature that triggers afferent nerve activity towards the central nervous system, which as a result produces efferent sympathetic outflow towards various organs, including resistance vasculature, heart and kidneys (Figure 1). The idea of RDN is supported by this knowledge of the pathophysiology.
Sympathetic overactivity as therapeutic target
There is abundant evidence clearly linking sympathetic hyperactivity with poor clinical outcomes in hypertensive patients, in chronic kidney disease and in heart failure patients, including higher left ventricular mass, vascular hypertrophy, kidney failure progression and risk of cardiovascular mortality as discussed in more detail elsewhere20,29. A more detailed discussion is out of the scope of this brief review.
Are there reasons to believe that present-day treatment affects sympathetic activity? The answer is yes.
In the late 1990s, we tested the hypothesis that an ACE inhibitor would reduce MSNA in CKD patients. Indeed, we found that enalapril effectively reduces MSNA in CKD patients. In contrast, amlodipine, albeit an effective blood lowering agent, increased MSNA30. Subsequent studies showed that Ang II receptor antagonists and renin inhibitors reduced MSNA by the same amount as ACE inhibitors31-33. The precise mechanism as to how RAAS inhibitors reduce MSNA is not clear. Our hypothesis is that effects on the kidney level are the most important. However, we cannot exclude an effect on the level of the central nervous system as well. Also, in essential hypertension and heart failure there is clear evidence that RAAS inhibitors reduce MSNA20.
Do sympatholytic drugs have the same effect? In part, yes. The central sympatholytic drug moxonidine reduces sympathetic activity in all stages of kidney failure34,35. Old studies have shown that some beta-blockers may reduce MSNA whereas others do not36,37. Diuretics usually increase MSNA with the exception of spironolactone37-39.
So, it can be concluded that RAAS inhibitors reduce sympathetic activity most likely by their effect on kidney level. Other antihypertensive agents have variable effects.
RENAL DENERVATION
If we accept the pathophysiologic mechanisms outlined above, RDN seems a logical next step and therefore an important new treatment option. Indeed, in the previous century splanchnicectomy was introduced as a treatment for malignant hypertension. Enormous blood pressure reductions could be obtained and the five-year mortality rate went down from 99% to 66%. However, there were various serious side effects, and this approach was abandoned once pharmacological treatment was introduced in the 1950s and 1960s40.
Recent technological advances make it possible to perform catheter-based RDN. Indeed, first results in patients with so-called resistant hypertension suggest that a substantial blood pressure drop can be obtained by this procedure41. The precise mechanism of this blood pressure lowering effect is still unclear.
Given the pathophysiological mechanisms outlined above, several effects could be expected, when the function of efferent nerves were disrupted. Recently, a small decrease in renal vascular resistance together with an improvement in central haemodynamics was reported in hypertensive subjects42,43. Some very preliminary data suggest a decrease in renin activity10. Clearly, more studies need to be done in order to find out to what extent disruption of efferent nerves contributes to the overall effect of RDN. Further, it seems especially interesting to investigate whether renal sodium handling changes as a result of the intervention.
What evidence is there for disruption of the afferent nerves? One would expect a decrease in total peripheral resistance as an important contributor to the blood pressure decrease as a result of RDN. However, no such data exist. The early observation that RDN also reduces MSNA is of great significance10. In this report of a single patient, MSNA was reduced by more than 50%. The fact that this effect did indeed occur can best be explained by accepting that the function of renal afferent nerves is disrupted. Also the magnitude of the effect is exceptional, because there is no pharmacological intervention that causes a decrease in MSNA of this size. Later reports were clearly less optimistic in this respect44,45. The obvious conclusion is that more research needs to be done.
Indeed, a decrease in left ventricular mass was reported in one study46. To what extent this is caused by blood pressure reduction per se or in sympathetic activity is difficult to distinguish. In one study an improvement in insulin sensitivity was reported47. This is a very interesting and potentially relevant observation. It supports the idea that disruption of the afferent nerves is important but it also sheds some light on the interaction between insulin and sympathetic activity. It suggests that high sympathetic activity causes a decrease in insulin sensitivity rather than the other way around. Therefore, it seems worth exploring this option further in obese/type II diabetes patients.
Altogether, there is some clinical evidence that RDN indeed disrupts the function of afferent nerves.
Given these considerations, the question arises what type of patient is likely to benefit most from the procedure? It seems safe to hypothesise that patients in whom it can be expected that the above-mentioned pathophysiological mechanisms are operational are likely to benefit most. General features of these patients include kidney failure/kidney injury and/or high central sympathetic outflow. These include patients with hypertension, but especially also those with chronic kidney disease and heart failure.
An often heard logical question is whether we actually need the renal nerves. It seems unlikely that renal nerves exist without a reason. In other words, are detrimental effects to be expected from destroying the nerves? From an evolutionary point of view, renal nerves seemed handy to have, when situations of extreme fight or flight were common. Nowadays, these situations are less likely to occur. Further, from decades of experience with kidney transplantation, in which the transplanted kidney is by definition totally denervated, we know that the absence of renal nerves does not seem to cause any serious problem to the transplanted patient. Finally, it seems very unlikely that the catheter-based RDN procedure results in 100% destruction of all nerves in the renal artery.
Conclusion
We can conclude that we stand at the beginning of a new era. There is convincing experimental and clinical evidence that targeting the renal nerves makes sense. It seems fair to conclude that the afferent nerves especially are involved in the pathophysiology of various disease conditions. Catheter-based RDN is a very interesting new treatment option, which could be of great value to numerous patients. First results are promising, but many questions are unanswered. Given the great potential, there is a great need for good research. It is clear that in order to do that effectively interventionists and referral physicians should work closely together.
Funding
R.L. de Jager is supported by a grant from The Netherlands Organisation for Health Research and Development (ZonMw grant 837004006).
Conflict of interest statement
R. L. de Jager has no conflicts of interest to declare. P. J. Blankestijn receives grants from Medtronic, the Dutch Kidney Foundation, and The Netherlands Organisation for Health Research and Development, and speaker and consultancy fees from Medtronic and St. Jude Medical.
