- in-stent restenosis
- drug-eluting balloon
- everolimus-eluting stent
- optical coherence tomography
Abstract
Background: The optimal treatment of bare metal stent restenosis is still not defined. The most employed contemporary option is the implantation of a drug-eluting stent (DES). However, this procedure implies the addition of a second metal layer in the vessel wall, which is linked to delayed healing. Furthermore, there may be a increased risk of malapposition of both struts of the bare metal and the newly implanted drug-eluting stent. These phenomena may give rise to an increased risk of stent thrombosis in this patient population. Recently, drug-eluting balloons (DEB) have been proposed as a new treatment strategy for bare metal stent restenosis. The initial results of this technique look promising.
Objectives: To compare healing processes after treatment of bare metal stent (BMS) in-stent restenosis (ISR) with balloon dilatation using DEB versus implantation of DES.
Study design: This is a prospective, multicentre (University Hospitals Leuven and ZOL Hospital Genk, Belgium) randomised clinical trial with clinical, angiographic and OCT follow-up at nine months. Patients with bare metal stent restenosis and an indication for repeat PCI are randomised to treatment with a paclitaxel-eluting balloon (SeQuent Please, B-Braun, Melsungen, Germany) versus a Xience V/ Xience Prime everolimus-eluting stent (Abbott Vascular, Santa Clara, CA, USA). The primary objective of this study is to evaluate the vascular healing response of the vessel wall after balloon angioplasty with a paclitaxel-eluting balloon versus implantation of a drug-eluting stent in patients with in-stent restenosis in a coronary artery. The primary endpoint of the study is stent strut coverage and stent strut apposition at nine months, as assessed with OCT.
Conclusions: Currently no prospectively collected data on vessel wall healing after treatment of in-stent restenosis, whether with DES or with DEB, are available. Therefore, the SEDUCE trial will yield pivotal insights on this important topic and guide further optimisation of the interventional treatment for this condition.
Summary
The optimal treatment of bare metal stent restenosis is still not defined. The most often employed contemporary option is the implantation of a drug-eluting stent (DES). Recently, drug-eluting balloons (DEB) have been suggested as a valuable alternative treatment strategy. The SEDUCE randomised controlled trial aims at comparing healing processes after treatment of in-stent restenosis with balloon dilatation using DEB versus implantation of DES, using intracoronary optical coherence tomography.
Introduction
In a meta-analysis of randomised trials comparing the effectiveness of drug-eluting stents versus balloon angioplasty or vascular brachytherapy for patients with bare metal in-stent restenosis (ISR), DES appeared to be markedly superior with reduced rates of target lesion revascularisation and of angiographic restenosis1. This has led to wide adoption of DES for ISR. Being an off-label indication, however, this might imply inherent safety risks associated with delayed healing. Recently, drug-eluting balloons (DEB) were proposed as a new treatment strategy for bare metal stent restenosis. In the PEPCAD II ISR (Paclitaxel-Eluting PTCA-Balloon Catheter in Coronary Artery Disease-2 In-Stent Restenosis)2, treatment of ISR using a paclitaxel-coated balloon resulted in better six-month angiographic results, than stenting with the Taxus (Boston Scientific, Natick, MA, USA) stent with a reduced late lumen loss (cut by about half). There was also a significant decrease in the rate of binary restenosis (7% vs. 30%) and superior six-month rates of TLR (6.3% vs. 19.4%) and MACE (7.8% vs. 16.9%). The results of this trial have led to a modification in the revascularisation guidelines, which recommend that drug-eluting balloons should be considered for the treatment of in-stent restenosis after prior bare metal stent (BMS)3. The main difference between coated balloons and DES in treating ISR is that after dilatation, no additional stent is left behind. Also, the paclitaxel coating extends over the entire length of the DEB and is distributed more evenly to the arterial wall. The drug-coated balloon delivers an initially homogeneous drug concentration to the arterial wall, which has been shown to be an effective substitute for sustained release4. If larger trials indeed prove that these devices to be as effective as DES (or even more so) in the treatment of ISR, they offer several possible advantages over the use of DES: no extra layer of metal implanted (avoiding the reduction of the flexibility of the vessel and the limitation of the repeatability of the procedure), easier deliverability to the vessel wall, need for only a short duration of clopidogrel therapy, and lower cost. At this time, however, no data exist on the effect of both treatment options to the vessel wall, specifically regarding stent strut coverage and stent strut apposition.
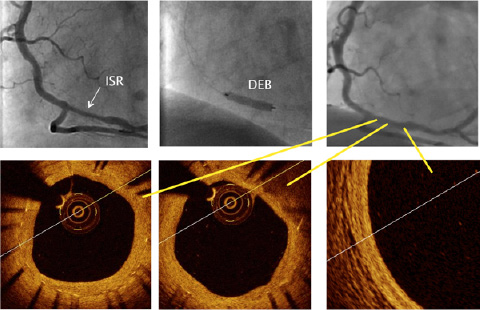
Figure 1. DEB for BMS ISR. A) BMS ISR in the distal RCA, B) after lesion pretreatment, application of a DEB, C) angiographic appearance at 9-months, D and E) moderate neointima growth at the treated segment, F) normal aspect of intima/media/adventitia just distal to the treated segment.
Optical coherence tomography (OCT) is a new imaging modality that visualises the intracoronary features with high axial resolution (10-20 µm); which is far better than the resolution of IVUS being around 100-150 µm. This extremely high resolution allows for characterisation and quantification of neointimal coverage of stent struts and for vastly improved appreciation of stent strut apposition. This technology also allows for the first time the in vivo assessment of vessel wall healing. The pivotal role of vessel wall healing in the pathogenesis of stent thrombosis has been beautifully illustrated in several recent publications5,6. It is likely that optical coherence tomography (OCT) will play an important role in the treatment of ISR in the near future, especially to evaluate the pro’s and con’s of DEB use. This technology is able to provide a very detailed assessment of the restenosed coronary artery segment and the result of its treatment, both in the acute phase and at follow-up. Several studies have pointed to a widely heterogeneous clinical presentation of patients with ISR, ranging from acute infarction to silent occlusion7. The same variability holds true for the angiographic and OCT appearance of ISR lesions8. The latter is especially relevant for drug-eluting stent (DES) restenosis, who often have highly variable imaging characteristics (heterogeneous versus homogeneous restenotic tissue, smooth versus irregular contours, with and without intraluminal structures…). These varying imaging characteristics have been shown to correlate with the clinical presentation of the patient (stable versus unstable coronary syndromes), as described by Gonzalo et al9. Recently, lipid-rich plaque with a thin fibrous cap in late bare metal stent (BMS) restenosis, a formerly unknown entity, was described using OCT10,11. In addition, adequate pretreatment of the lesion is deemed critical for the long-term result of ISR lesions, whether treated with drug-eluting stent implantation or application of a drug-eluting balloon. Especially in the latter, the inflation of aDEB at the site of the restenotic segment is only intended as a local delivery of the antiproliferative drug. Hence, all measures of correcting stent malapposition and underexpansion and the mechanical removal of the great bulk of neointimal tissue should be done before. The ease of use and the safety of the Fourier domain OCT imaging system (FD-OCT), with its rapid exchange introduction system and its ultra-fast pullback (20 mm/s), facilitate assessment of every step taken treating an ISR lesion (including the effect of non-compliant, scoring and cutting balloons) and assist the operator to decide at which point no better “preparation” of the lesion can be achieved12. After application of a DES or a DEB, afinal OCT pullback can serve as a final check to verify if any additional interventions (e.g., for edge dissection or other mechanical problems) are necessary. This final pullback also serves as an ideal baseline comparator for longer-term follow-up. In fact, the combination of baseline OCT images of a restenotic lesion, combined with those directly after treatment and at mid-term follow-up or at the time of treatment failure, will provide invaluable information on guiding patient treatment and device development in the future.
Study design
This is a prospective, multicentre (University Hospitals Leuven and ZOL hospital Genk, Belgium) randomised clinical trial with clinical, angiographic and OCT follow-up at 9-months. Patients with bare metal stent restenosis for which a repeat PCI procedure is scheduled, are randomised to treatment with a paclitaxel-eluting balloon (SeQuent Please, B-Braun, Melsungen, Germany) versus aXience V/Prime everolimus-eluting stent (Abbott Vascular, Santa Clara, CA, USA). The primary objective of this study is to evaluate the vascular healing response of the vessel wall after balloon angioplasty with a paclitaxel- eluting balloon versus implantation of a drug-eluting stent in patients with in-stent restenosis in a coronary artery.
Study population
The study population will consist of 50 patients presenting with bare metal in-stent restenosis. In patients presenting with symptoms of angina or documented ischaemia, and in whom an in-stent restenotic lesion is deemed to be the culprit lesion, informed consent for participation in the study is obtained. Inclusion and exclusion criteria are detailed in Table1. There is no exclusion based on the type of BMS implanted.
| Table 1. SEDUCE inclusion and exclusion criteria. |
| Inclusion criteria |
| 1.Patients older than 18 years |
| 2.Written informed consent available |
| 3.Patients eligible for percutaneous coronary intervention |
| 4.Patients with a single or multiple restenotic lesion(s) in a previously bare metal stented area of a coronary artery |
| 5.Target reference vessel diameter measured by QCA: 2-4 mm |
| 6.Target lesion length measured by QCA <24 mm |
| 7.Target lesion stenosis measured by QCA: >70%- <100% |
| 8.Patients willing to provide written informed consent prior to participation and willing and able to participate in all follow-up evaluations |
| Exclusion criteria |
| 1.Left ventricular ejection fraction of <30% |
| 2.Impaired renal function (serum creatinine >2.0 mg/dl) |
| 3.Target lesion located in bifurcation |
| 4.Previous and/or planned brachytherapy of target vessel |
| 5.Lesion of the left main trunk >50%, unprotected |
| 6.Known allergies to antiplatelet, anticoagulation therapy, contrast media, paclitaxel or everolimus |
| 7.Pregnant and/or breast-feeding females or females who intend to become pregnant (pregnancy test required) |
| 8.Patients with a life expectancy of less than one year |
| 9.Patients who intend to have a major surgical intervention within 6-months of enrolment in the study. |
| 10.Patients currently enrolled in other investigational device or drug trials |
| 11.Patients not able or willing to adhere to follow-up visits |
| 12.Patients who previously participated in this study |
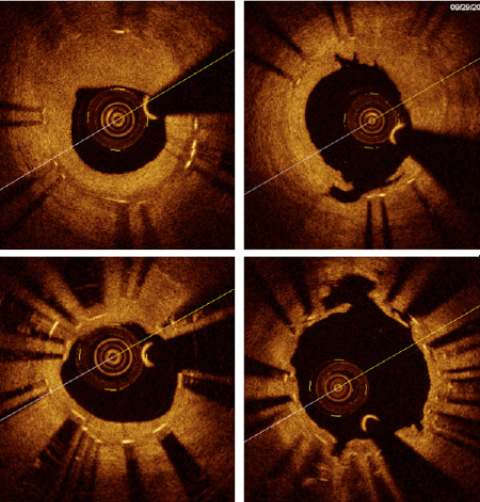
Figure 2. Lesion preparation in ISR. A) Bare metal in-stent restenosis with severe neointimal hyperplasia. B) Result after treatment with anon-compliant balloon at high pressure and a cutting balloon (neointimal tears at 12 o’clock). C) Stent underexpansion in a case of re-restenosis in a DES for BMS ISR. There is an uncovered strut at 4 o’clock. D) Result after multiple dilations with non-compliant balloons at high pressure.
Study procedure and randomisation
The procedure starts as a conventional PCI procedure. The in-stent restenotic lesion is crossed with a conventional coronary guidewire. Then, extensive predilation of the lesion is performed. Several balloons can be used, if desirable, but the predilation procedure ends with a non-compliant balloon, sized according to vessel size, at high inflation pressure (16-20 atm). After predilation, the angiographic result is carefully assessed by the operator. Only if the operator judges that an acceptable angiographic result is obtained and that –for angiographic reasons– no strict indication for stent implantation is present, the randomisation procedure is started by site study personnel. If an important dissection or recoil with insufficient lumen gain after predilation occur, the patient is not included in the study. Patients meeting the eligibility criteria are subsequently randomised in the order they qualify. Patients are considered enrolled in the study and eligible for the final intention to treat analysis at the time of randomisation. The randomisation process is organised via an independent centre (LCC, Leuven Coordinating Centre) for both sites, using an IVRS. For practical reasons, the operator and the patient are not blinded to the allocated treatment group.
After randomisation, the randomised treatment strategy is applied. In the case of randomisation to the drug-eluting balloon, the size of the drug-eluting balloon is chosen at least 1:1 to the reference vessel size, and the length in order to achieve an overlap of at least 3mm at both edges of the stent. The drug-eluting balloon is inflated for 60 seconds and then retrieved from the coronary artery. The result is checked angiographically, and if no substantial problems (e.g., edge dissection) are discovered, the procedure is terminated. In case of drug-eluting stent implantation, the appropriately sized stent is inflated at high pressure (14-18atm) for at least 20 seconds. If deemed necessary, additional non-compliant balloon post-dilation at high pressure or additional stent implantation can be performed.
Following the intervention, a final angiogram will be performed to document the residual lumen diameter and the vessel diameter. Intracoronary nitroglycerin (100-200mcg) is be given immediately prior to the final angiography. The final angiography should be obtained repeating the two near orthogonal views that reveal the target lesion free of foreshortening or vessel overlap. All details of the angiographic procedure are recorded.
Study follow-up
Patients have follow-up at the outpatient clinic at one and eight months after inclusion. Angiographic follow-up, with optical coherence tomography (OCT) examination of the treated segment, is performed at 9-months after inclusion. A yearly clinical follow-up at 1, 2, 3, 4 and 5 years is prespecified by protocol.
For the 9-month OCT control examination, a standardised protocol is applied in both participating centres. Both centres use the C7XR Fourier domain OCT imaging console (St. Jude Medical, St. Paul, MN, USA) and the corresponding Dragonfly Intravascular Imaging Catheter. Intracoronary injection of nitrates (0.1 to 0.3mg nitroglycerine) is performed prior to insertion of the imaging catheter. In order to improve the accuracy and reproducibility of the OCT assessments, meticulous attention is paid to achieve optimal blood clearance of the coronary artery, via flushing with contrast medium using an automated pump. The lens of the OCT wire is placed at least 5mm distal to the distal edge of the lesion and the automatic pull-back through the lesion is performed including at least 5 mm of the proximal and distal vessel.
Primary and secondary endpoints
The primary endpoint is the percentage of uncovered struts at nine month follow-up, as assessed with OCT. The secondary endpoints are stent apposition at nine months, assessed with OCT, Late lumen loss (in-stent), binary in-stent restenosis and binary in-segment restenosis, minimal lumen diameter (MLD), in-stent and in-segment at nine months, absolute and percent volume of intimal hyperplasia at nine months measured with OCT, cumulative MACE rate at 12 months. MACE is defined as a combination of events including cardiac death, non-fatal MI, clinically driven TLR, TVR, emergent coronary artery bypass surgery (CABG), stent thrombosis at all follow-up.
Ethics and informed consent
The study will be performed in accordance with the applicable regulations, the declaration of Helsinki and Good Clinical Practice. The ethics committee at both study sites approved the study protocol.
Statistical analysis
Due to the exploratory nature of this study, no formal sample size calculations were performed. Instead, the sample size of 25 patients per group is largely driven by practical considerations. All data collected in this study will be summarised and presented by randomised group. Continuous data will be summarised by their mean, standard deviation, minimum, Q1, median, Q3 and maximum. Categorical data will be presented by their frequency and percentage. All statistical tests will be 2-sided and assessed at the 5% significance level.
For the analysis of the primary endpoints, a 2-sample Wilcoxon rank-sum test will be used. The treatment difference will be quantified using the Hodges-Lehmann estimator, which will be presented along with its 95% confidence interval. Statistical significance will be assessed at the 5% significance level. Additional analyses of the primary endpoints will be done whereby both endpoints will be dichotomised using a 5% and a 10% cutoff. Comparisons between groups will be made using the continuity-corrected chi-square test. The relative risk between treatment groups will be estimated and presented along with its 95% confidence interval.
For the secondary endpoints, continuous, normally distributed values will be analysed using an analysis of variance (ANOVA), containing factors for group and randomised treatment. Treatment differences will be estimated and presented along with their 95% confidence interval. Continuous, non-normal and binary secondary endpoints will be analysed as described for the primary endpoints. The cumulative incidence of MACE will be assessed using Kaplan-Meier curves and comparisons between the treatment groups will be made using a stratified log-rank test, with group as stratification factor. All analyses of the primary and secondary endpoints will be done according to the intention to treat (ITT) principle, i.e., all randomised patients will be included into the analysis according to their randomised treatment.
Study organisation
The SEDUCE study is an investigator-initiated study, thanks to grant number G.0690.09 from the Research Foundation Flanders. The investigators are supported by Abbott Vascular through aresearch grant and by B-Braun in the form of supply of drug-eluting balloons in the frame of the study. The authors are solely responsible for the design and conduct of this study, all study analyses, the drafting and editing of the manuscript and its final contents.
Current progress
First enrolment of patients started in June 2009. End of January 2011, 32/50 patients were included and 9-month angiographic and OCT follow-up was obtained in 17. Complete enrolment is expected by the end of 2011 and final results to be presented at the end of 2012.
Conflict of interest statement
Tom Adriaenssens has received speakers fees from St Jude Medical and a research grant from Abbott Vascular. He is supported for this study by B-Braun in the form of supply of SeQuent Please drug-eluting balloon catheters. The SEDUCE study is sponsored by the Research Foundation Flanders through an unrestricted grant.
Walter Desmet received a research grant form Abbott Vascular.
The other authors report no conflict of interest.
