
Abstract
Aims: The global, prospective AMPLATZER Amulet observational study documents real-world periprocedural, transoesophageal echocardiographic (TEE) and clinical outcomes from left atrial appendage occlusion (LAAO) using the AMPLATZER Amulet device. The aim of this report is to describe the periprocedural and early clinical/TEE results from this study.
Methods and results: This multicentre prospective real-world registry included 1,088 patients (75±8.5 years, 64.5% male, CHA2DS2-VASc: 4.2±1.6, HAS-BLED: 3.3±1.1) with non-valvular atrial fibrillation; 82.8% of patients were considered to have an absolute or relative contraindication to long-term anticoagulation and 72.4% had had a previous major bleeding. Periprocedural results, clinical outcomes up to the first three months and the available TEE results from the first scheduled follow-up (one to three months post implant) are reported. Successful device implantation was achieved in 99.0% of patients. During the procedure and index hospitalisation, major adverse events occurred in 3.2% of patients. Patients were discharged on a single antiplatelet agent (23.0%), dual antiplatelets (54.3%) or an oral anticoagulant (18.9%). TEE follow-up 67±23 days post procedure in 673 patients showed adequate (<3 mm jet) occlusion of the appendage in 98.2% of patients and device thrombus in 10 patients (1.5%), as evaluated by core laboratory analysis.
Conclusions: This large real-world prospective registry of catheter-based LAAO using the AMPLATZER Amulet device reports a high implant success rate and a low periprocedural complication rate in a population with a high risk of stroke and bleeding. Transoesophageal echo data confirm good closure rates during follow-up and low rates of device-associated thrombus.
Abbreviations
AF: atrial fibrillation
APT: antiplatelet therapy
CEC: clinical events committee
LAA: left atrial appendage
LAAO: left atrial appendage occlusion
MAE: major adverse events
OAC: oral anticoagulation
SADE: serious adverse device effects
TEE: transoesophageal echocardiography
TIA: transient ischaemic attack
TTE: transthoracic echocardiography
Introduction
Percutaneous, catheter-based occlusion of the left atrial appendage (LAA) is an increasingly performed procedure aiming to reduce the risk of stroke in patients with non-valvular atrial fibrillation (AF)1-3. Randomised clinical trials1,2 have suggested that percutaneous LAA occlusion (LAAO) can represent an alternative to anticoagulation in warfarin-eligible patients. However, by the elimination of OAC-associated bleeding, it is potentially a particularly attractive option for patients with contraindications to the use of oral anticoagulants (OAC) due to their high bleeding risk. While the implantation of an LAAO device may be associated with upfront periprocedural risks, studies have shown a gradual reduction of these risks over time, as operators gain more experience with the therapy4. Recently, a large retrospective multicentre study in patients receiving the AMPLATZER™ Cardiac Plug (ACP) (St. Jude Medical [now Abbott], St. Paul, MN, USA) device and a large real-world registry using the WATCHMAN™ device (Boston Scientific, Marlborough, MA, USA) have reported favourable outcomes of LAAO in all-comers populations3,5,6. These populations particularly included many patients with a contraindication to long-term OAC, whereas, by design, randomised trials had to include patients eligible for OAC1,2. As expressed by the EHRA/EAPCI expert consensus statement, LAAO can be considered for AF patients at risk of ischaemic stroke who have an unacceptably high bleeding risk, refuse anticoagulant therapy or are contraindicated for systemic anticoagulation7.
Currently, the WATCHMAN device and the AMPLATZER™ Amulet™ device (St. Jude Medical) are the most commonly implanted devices for catheter-based LAAO, with a higher penetration of the Amulet device within Europe compared with non-European geographies8. The Amulet device is a second-generation LAAO device, based on the ACP device and incorporating several incremental design improvements. A few relatively small series have been reported with this device9-11. Comparative studies have shown similar results obtained with the first- and second-generation AMPLATZER devices in terms of safety, implantation success and appropriate closure of the LAA12,13. An FDA approval trial for this device is currently ongoing, with the aim of collecting randomised controlled data from the Amulet and WATCHMAN devices from 1,600 patients worldwide14.
Here, we present the procedural, early clinical and TEE outcomes of a large global real-world prospective registry including an all-comers population and using the AMPLATZER Amulet device. In this global registry, serious adverse events were adjudicated by an independent clinical events committee and echocardiographic images were evaluated by an independent core laboratory.
Methods
This prospective, global, observational study documents periprocedural, transoesophageal echocardiographic (TEE) and clinical outcome data on catheter-based LAAO using the AMPLATZER Amulet device for ischaemic stroke prevention in patients with non-valvular AF. The present study summarises the results of periprocedural outcomes (within seven days of implant) and early clinical and TEE follow-up data up to three months from implant.
The study adhered to international rules for scientific studies, the Helsinki principles, with local ethics committee approval in participating centres. Patients provided informed consent prior to the procedure. St. Jude Medical/Abbott provided funding for the study.
PATIENTS
The study included 1,088 patients with paroxysmal, persistent or long-standing persistent non-valvular AF, enrolled in 61 centres in Europe, Australia, Israel, Chile and Hong Kong. To be included in the study, patients were required to be eligible for percutaneous LAAO according to current standard indications per judgements of the institutional treatment team. Patients were considered enrolled in the study after they had consented and the dilator/delivery system had been introduced into the vasculature. Exclusion criteria included any evidence of intracardiac thrombus, active infection or endocarditis or other infections producing bacteraemia, anticipated device interference with intracardiac or intravascular structures, medical disorders expected to interfere with the planned clinical assessments and LAA anatomy not accommodating a device according to sizing guidelines. The risks of ischaemic stroke and bleeding were assessed by the CHA2DS2-VASc15 and HAS-BLED16 scores, respectively.
LAAO PROCEDURE
The LAAO procedures were performed following local institutional routines and procedures and were consistent with the manufacturer’s recommendations. Participating investigators were required to have a minimum LAAO implanting experience with the study device, including three to five proctored cases and five cases as the principal implanting physician. Given the real-world nature of the registry, the level of experience with the Amulet device and overall implant volume varied among the participating sites. Five percent of the study centres perform more than 100 LAAO procedures/year. Prior to commencing the actual implantation procedure, imaging was performed to exclude left atrial (LA) and LAA thrombus, and to evaluate the relevant anatomy and dimensional information required for device sizing. Devices were implanted under imaging guidance, including fluoroscopy, angiography and TEE or intracardiac echocardiography (ICE). Device implantation was considered as starting upon crossing the interatrial septum.
This registry was conducted to assess the safety and clinical outcome of LAAO using the AMPLATZER Amulet device. This second-generation LAAO device (Figure 1) is an incremental development based on the first-generation ACP device. The device has a proximal disc to seal the ostium of the LAA and a distal lobe, to be positioned within the LAA. Both the disc and the lobe are constructed from nitinol mesh and incorporate an internal fabric to promote intra-device occlusion. Retention wires on the lobe facilitate anchoring and stabilisation of the device. The device is provided pre-loaded within its dedicated delivery sheath. The device is described in detail elsewhere17.
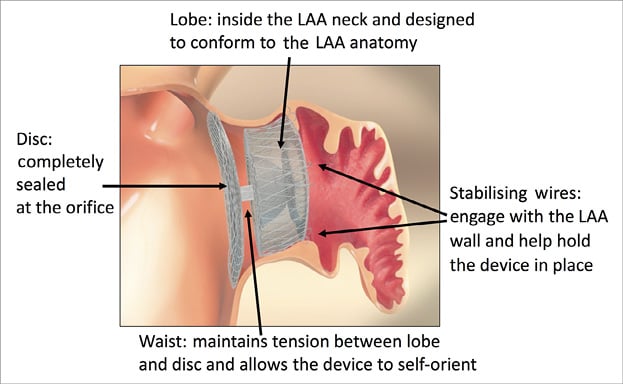
Figure 1. AMPLATZER Amulet left atrial appendage occluder.
FOLLOW-UP
Following the device implantation, a first follow-up visit was scheduled within a one- to three-month window after the procedure. Adverse events during the follow-up period were reported by the study sites independently from scheduled study visits. Patients in whom device implantation was commenced but who eventually did not receive a device were followed for seven days after the procedure in order to document the occurrence of any adverse events. Here we report the periprocedural results (within seven days of implant), early TEE outcomes available from the first scheduled study visit as analysed by the core laboratory, and adverse events reported during the three-month post-implant period.
SAFETY EVENTS
Figure 2 provides a graphical representation of the timing of the two adverse event metrics, including major adverse events (MAE) and serious adverse device effects (SADE), used in the study. A primary safety assessment involved MAE during the procedure and index hospitalisation, defined similarly to the Munich expert consensus document on LAAO studies and endpoints18. This assessment was performed to characterise the procedural safety and success. MAE included death, stroke, embolism, bleeding of Bleeding Academic Research Consortium (BARC) type 3 or higher19, all device embolisations and major vascular complications. In contrast to other studies that report on safety events, not only pericardial effusions or tamponades requiring surgery were considered MAE, but also those that were addressed through pericardial drain. All device embolisations, including those requiring percutaneous or surgical retrieval, were considered MAE. Additionally, the safety events of this study were characterised following a regulatory definition (consistent with ISO 14155 and the MEDDEV 2.7/3) of SADE which were assessed during the procedure up to seven days post procedure (periprocedural) and >7 days after the procedure (late events) up to three months. Additional endpoints included the composite of ischaemic stroke, systemic embolism and cardiovascular death, and bleeding events. An ischaemic stroke was defined as an acute episode of focal cerebral, spinal or retinal dysfunction caused by infarction of the central nervous system tissue. Diagnosis of stroke or transient ischaemic attack (TIA) was based on confirmative neurological evaluation and brain imaging. To minimise bias and subjective interpretation, serious events were adjudicated by an independent clinical events committee (CEC), including clinical experts experienced in relevant clinical specialties and not actively involved in the conduct of the study (Appendix). Events were adjudicated as device- and/or procedure-related if a causal relationship was suspected with the LAAO device and/or the delivery system and/or the procedure. Additional outcome measures included technical and procedural success. The use of oral anticoagulants (OAC) and/or antiplatelet therapy (APT) during the follow-up period was evaluated. Technical success was defined as the successful implantation of the device in the correct position. Procedural success was defined as technical success without MAE during the procedure and index hospitalisation.
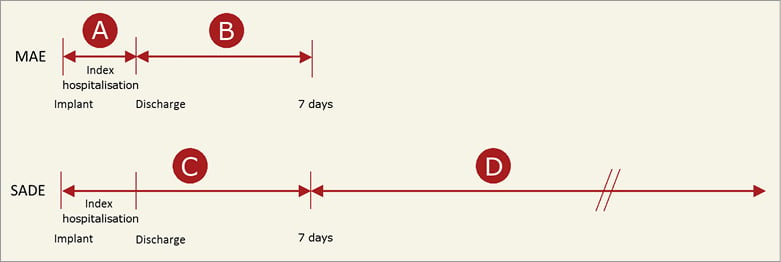
Figure 2. Timing of the adverse event metrics (MAE: major adverse events, and SADE: serious adverse device effects). The implantation procedure is included within periods A and C. For some patients, discharge was >7 days after the implantation procedure, in which case periods A and C were extended to include the complete index hospitalisation.
IMAGING
Imaging was used to assess patient eligibility for device implantation, to guide the implantation procedure and to evaluate the implanted device during follow-up. Device implantation was most commonly guided by TEE, while transthoracic echocardiography (TTE) was used at discharge to assess any pericardial effusion or identify possible device embolisation. In addition, TEE was performed at the first follow-up visit at one to three months after implantation to assess peri-device leak and device-related thrombus. TEE was also performed in case of a confirmed stroke or TIA to determine the association with device-related thrombus. All echocardiographic images were evaluated with regard to peri-device leak and device-related thrombus by an independent core laboratory (MedStar Health Research Institute, Hyattsville, MD, USA). Thrombus on the device was defined as the presence of an echogenic mass attached to the LA side of the device, identified in at least two different views. Residual leak was identified as flow around the device lobe within the LAA detected by colour flow Doppler echocardiography. To evaluate the degree of leak, the neck of the jet was measured on a perpendicular axis to its direction. Peri-device leak was defined as absent, small (<3 mm jet), medium (3 to 5 mm) or large (>5 mm). LAA closure was considered clinically adequate if a leak was either absent or less than 3 mm.
DATA ANALYSIS
The present study involves evaluation of the implantation procedure and periprocedural outcomes, as well as early TEE follow-up data available from the first scheduled follow-up visit one to three months post implant and adverse events reported during the first three months after implantation. No formal hypothesis is being tested in this observational post-market study. However, point estimates and exact 95% confidence intervals are provided for the observed rates.
Results
PATIENTS
A total of 1,088 patients were enrolled between June 2015 and September 2016. Among the 61 participating centres, 34 centres enrolled >10 patients. The patients’ age was 75.2±8.5 years and 64.5% were male. Clinical characteristics of the patient cohort are shown in Table 1. The vast majority of patients had a history of major bleeding (72.4%) and contraindications to OAC (82.8%). With 64.9% of the patients having a CHA2DS2-VASc score ≥4 and 77.1% having a HAS-BLED score ≥3, the study cohort represented a population at high risk for stroke as well as for bleeding (Figure 3).
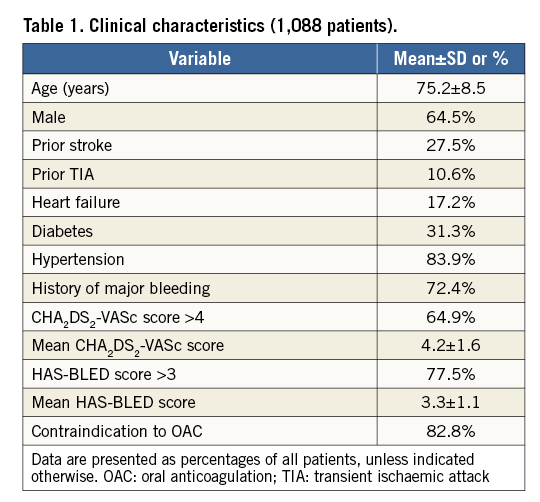
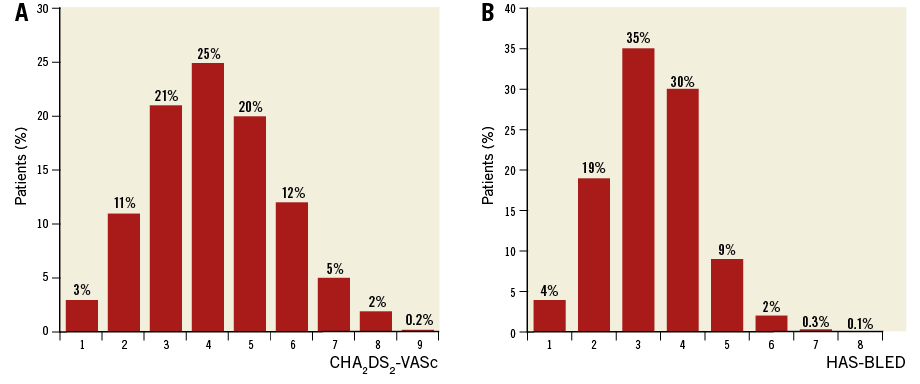
Figure 3. Distribution of stroke risk and bleeding risk in the study population, as assessed by the CHA2DS2-VASc (A) and HAS-BLED (B) scores.
DEVICE IMPLANTATION
Of the 1,088 patients enrolled, the Amulet device was successfully implanted in 1,077 patients, resulting in a technical success rate of 99.0%. Implantation was not successful in nine patients, most frequently due to the LAA anatomy. Anatomical reasons for implant failure included a shallow appendage or landing zone, a large diameter landing zone and incongruence between the septal puncture site and the LAA position. For two other patients, a medical decision was made to implant another LAAO device (one ACP and one WATCHMAN device) without attempting implantation of an Amulet device. Table 2 provides additional procedural details, including device sizes, number of devices per procedure and anaesthesia utilised.
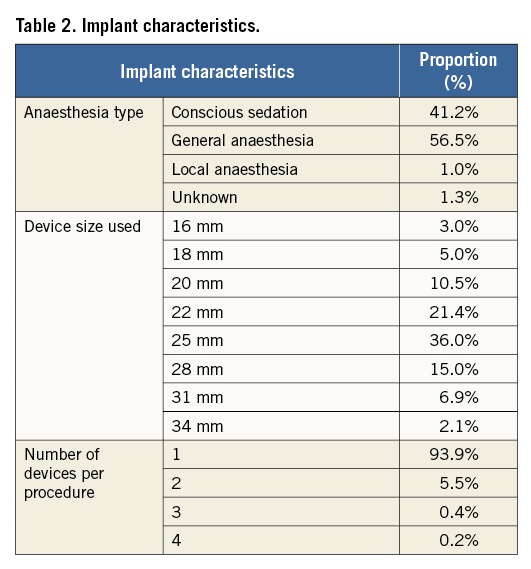
During the procedure and index hospitalisation, 35 patients experienced MAE (3.2%) (Table 3), with major bleeding being the most common event (26 events, 2.4%). Of these major bleeding events, 13 events (1.2%) were a result of pericardial effusion or cardiac tamponade, with 10 subjects (0.9%) requiring pericardiocentesis and three patients (0.3%) requiring surgery. Ten patients (0.9%) had a major vascular complication. Prior to discharge, two patients died due to cardiac perforation in one patient and cardiorespiratory arrest/shock in another patient. The patient with cardiorespiratory arrest underwent successful implantation with adequate LAAO and no signs of pericardial effusion. Intraoperatively, the patient experienced several episodes of hypotension, followed by recurrent hypotension and bradycardia at one day after the procedure. Eventually, the patient went into cardiorespiratory arrest and could not be resuscitated. The procedural success rate (technical success without MAE during the procedure and index hospitalisation) was 95.8%.
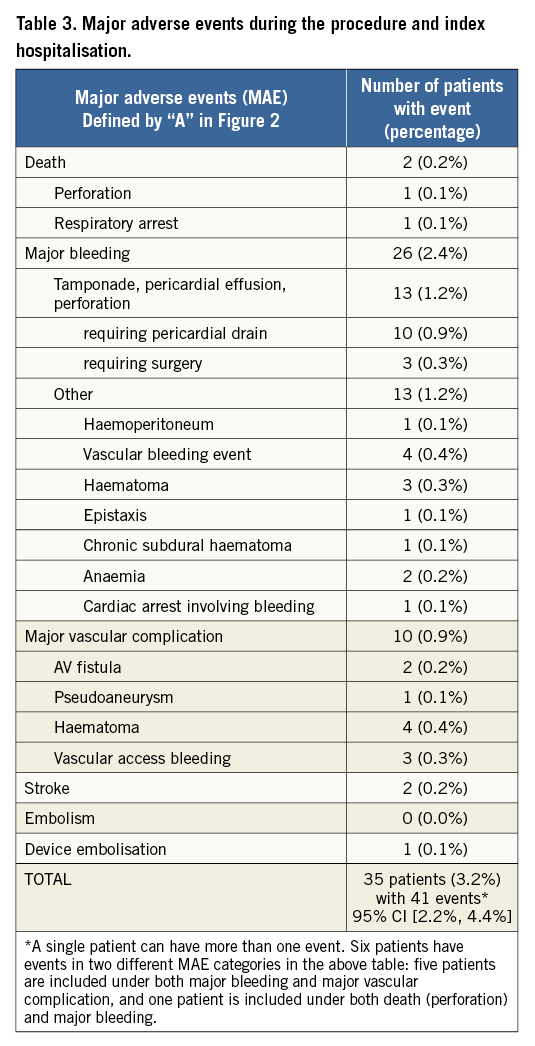
Further evaluation of adverse events included the assessment of MAE and SADE within seven days or during index hospitalisation, whichever was longer (Table 4). In addition to the MAE reported during the procedure and index hospitalisation, there were four other patients who had a major adverse event within seven days. One patient died due to myocardial infarction which was adjudicated as non-device-related. This patient underwent successful device implantation with adequate closure of the LAA and no pericardial effusion. The patient died two days after the procedure and autopsy revealed a fresh myocardial infarction of the left ventricle with stenosis secondary to coronary heart disease. A second patient was readmitted to the hospital due to chest pain within seven days of the procedure. Echocardiography revealed a pericardial effusion requiring pericardiocentesis. The two remaining events comprised gastrointestinal (GI) bleeding (one patient) and anaemia requiring blood transfusion (one patient). As a result, within seven days post procedure, MAE occurred in 39 patients (3.6%). Following regulatory definitions, there were 71 SADE in 61 patients within seven days of the implantation procedure or during index hospitalisation, representing a periprocedural SADE rate of 5.6%.
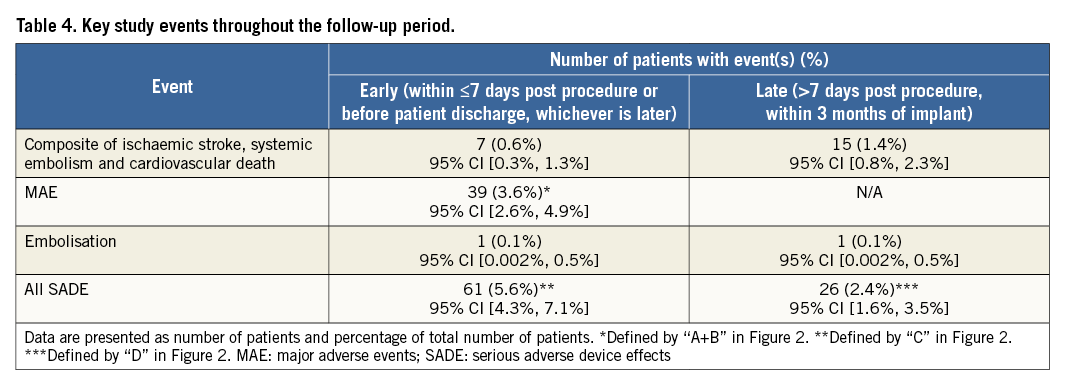
Within the seven-day post-procedural period, four ischaemic strokes occurred; in none of these cases was thrombus found on the device. Two strokes occurred on the day of the procedure and were both diagnosed as cardioembolic strokes with no signs of intraprocedural air embolism. The other two strokes occurred at two and six days after the procedure.
Within the entire implantation cohort, there were two cases of device-related thrombus within seven days of implantation. One case was identified in the patient with chest pain, discussed above, in whom echocardiography revealed pericardial effusion as well as thrombus on the device. In another patient, device thrombus was found during a transcatheter aortic valve implantation procedure, a few days after LAAO. Within the seven-day post-procedural period, there was one case of device embolisation, observed in a patient who became hypotensive, six hours post procedure. The device was successfully retrieved.
Intraprocedural TEE evaluation of the implant, as adjudicated by the core laboratory, showed adequate LAAO (i.e., peri-device leak <3 mm on colour flow Doppler) in 99.6% of the patients. At discharge, most patients (77.3%) were prescribed single or dual APT while 18.9% of the patients were on OAC. No antithrombotic therapy was prescribed for 2.0% of the patients (Table 5).

FOLLOW-UP
Per cut-off date, 23 patients died prior to the first follow-up visit, including 13 cardiovascular and 10 non-cardiovascular deaths. All remaining patients who were successfully implanted with the Amulet device reached their one- to three-month follow-up window. The one- to three-month follow-up visits were performed with a mean follow-up duration of 2.4±0.8 months. Table 4 provides an overview of key events reported up to three months after the procedure, which were reported independently of the scheduled follow-up visits. A total of 31 SADE in 26 patients occurred after seven days up to three months post procedure, representing a SADE rate of 2.4% after seven days post procedure.
Follow-up TEE results were available and evaluated by the core laboratory from 673 patients: these showed adequate (<3 mm) LAAO in 98.2% of patients. Overall, device-related thrombus was observed in 10 patients (1.5% of evaluated TEEs), which includes two patients with thrombus within seven days post implant and eight patients with detected thrombus more than seven days and up to three months after implantation. Of these 10 patients with device thrombus, three (30%) were on single and three (30%) on dual APT, and four (40%) were on OAC. These patients were treated with single APT (two cases), dual APT (one case) or OAC (seven cases). Among them, one patient had an ischaemic stroke. The device-related thrombus in this patient was observed at 43 days post implant and the stroke occurred at 71 days after implantation while the patient was on APT. Another device embolisation was identified during a scheduled follow-up visit at 90 days after the procedure. The device was percutaneously retrieved from the aortic arch and another device was implanted the next day without complications.
BLEEDING SERIOUS ADVERSE EVENTS (SAE) DURING THE FOLLOW-UP PERIOD
Between eight and 90 days post implantation, bleeding SAE of BARC type 3 or higher were reported from 43 patients (4.0%). The most frequent bleeding events, representing the majority of bleeding SAE during this time period, included GI bleeding (20 patients) and anaemia requiring blood transfusion (eight patients).
Discussion
This global prospective registry enrolled a large cohort of AF patients at high risk for ischaemic stroke as well as bleeding, implanted with the AMPLATZER Amulet device and assessed for periprocedural and early clinical and TEE outcomes. While data were collected from a real-world, multicentre cohort in an observational registry approach, measures to achieve consistent interpretation of the data included the evaluation of all echocardiographic images by a core laboratory and adverse event adjudication by an independent committee of clinical experts. Procedural results, reported here, indicate a high implantation success (99.0%) and adequate LAAO in almost all patients who received a device (99.8%). The clinically relevant metric of MAE during implantation and subsequent hospitalisation was 3.2%. It should be noted that the vast majority of patients in the study had a history of major bleeding and were considered relatively or absolutely contraindicated to OAC.
LAAO for the prevention of stroke in AF patients has been the subject of two randomised controlled trials and observational multicentre studies using the WATCHMAN device1,2,4,6. These studies have reported a gradual decrease in the rate of procedural complications over time, from 8.7% in the PROTECT AF trial to 4.2% in the PREVAIL trial2. Recently, the EWOLUTION registry reported a Kaplan-Meier estimate of the device- and/or procedure-related serious adverse event rate up to seven days after the procedure of 2.8%6. From a multicentre retrospective study using the ACP device, Tzikas et al3 reported a 4.97% rate of major periprocedural adverse events. Comparison of these procedural complication rates with those from the present prospective global AMPLATZER Amulet registry should be made with caution, given the variation in reporting routines and definitions. Both the EWOLUTION registry and the retrospective ACP registry relied on an investigator-reported relatedness of the events with the device and/or procedure. With the consistent adjudication of clinical events by an independent committee of clinical experts, the present global registry reports a rate of MAE during the procedure and index hospitalisation of 3.2%, which compares well with the serious adverse event rate assessed in the EWOLUTION registry and is lower than the rate reported by Tzikas et al from the ACP registry3.
Echocardiography was intensively used during the periprocedural and follow-up periods to identify device embolisation. Despite these rigorous inspections, device embolisation was very rare during the reported follow-up period. One embolisation occurred within seven days and another was observed at 90 days after implantation. These results are similar to those reported from the EWOLUTION registry6 (two device embolisations within 30 days after implantation), while the ACP registry3 reported one device embolisation requiring surgical removal and seven embolised devices that were retrieved percutaneously. Concerning this observation, the design modifications implemented in this second-generation LAAO device may have contributed to the relatively low embolisation rate.
Immediate adequate LAA closure, as assessed by periprocedural TEE, was achieved in 99.6% of the patients. This observed rate is similar to the immediate closure rate reported from the EWOLUTION registry (99.3%)6; however, the Amulet registry applied a more stringent criterion (peri-device leak <3 mm vs. ≤5 mm on colour Doppler) and implemented a consistent imaging evaluation protocol performed by an independent core laboratory. The rate of adequate LAA closure remained high in the assessed cohort up to the first follow-up (98.2%).
There was one ischaemic stroke throughout the reported follow-up period, in a patient who was identified as having a device-related thrombus. Overall, device-related thrombus was identified in two patients during the implantation procedure and in 10 patients during follow-up. Extended follow-up is required to evaluate this outcome versus the long-term rates reported from other studies (e.g., 4.2% and 4.4% from the PROTECT AF trial4 and the ACP registry3, respectively). The current data from this registry are insufficient to either confirm or deny a causal relationship between device-related thrombus and ischaemic stroke. This association may be difficult to demonstrate, given the low event rates. Per protocol, this ongoing registry will continue to verify device thrombus in patients with confirmed ischaemic stroke.
According to manufacturer recommendations, the WATCHMAN device requires OAC for a minimum of 45 days post implant, while antiplatelet therapy may be prescribed after implantation of the Amulet device. The antithrombotic therapy prescribed at discharge in this registry reflects an approach to reducing the level of antithrombotic medication after device implantation in patients at high risk of bleeding. As the majority of patients had a contraindication to OAC, anticoagulants were prescribed to only 18.9% of the patients.
Limitations
The reported study was conducted as a registry with its associated typical limitations. However, a registry enrolling an all-comer population is a well-accepted approach to collect real-world clinical data from a relatively large cohort. Several measures, which are not standard for a registry study, were implemented to ensure consistent data interpretation, including the involvement of a CEC and a core laboratory. The present analysis included results from follow-up TEE of approximately two thirds of the study cohort, which should be considered a limitation of this study. Nevertheless, the assessed population still represents a large core laboratory-evaluated echocardiographic database on LAAO. While we cannot exclude bias due to the fact that our analysis did not include TEE evaluation of all patients, we have no reason or indication to assume that the evaluated subcohort is not representative of our total all-comers population.
Conclusions
Compared with recently reported registries on LAAO, this study reported a high implant success rate (99%) and similar periprocedural risk (5.6% SADE and 3.2% MAE) from a real-world all-comers cohort of AF patients at high risk of stroke and bleeding. TEE follow-up data confirm high LAA closure rates (98.2%) at one to three months post implant. Follow-up of this cohort is ongoing to collect long-term clinical outcome data.
| Impact on daily practice Early results are presented from the ongoing global, prospective AMPLATZER Amulet observational study, including a large real-world cohort of AF patients undergoing LAAO with the second-generation AMPLATZER Amulet device. The cohort is characterised by a high risk for ischaemic stroke and major bleeding, while most patients were contraindicated to long-term anticoagulation. Results show successful and safe implantation within this real-world cohort (implantation success rate: 99%; rate of major adverse events during the procedure or index hospitalisation: 3.2%). TEE follow-up confirmed adequate occlusion of the LAA (98.2% of patients) and a low incidence of device-related thrombus (1.5%). |
Appendix. Clinical Events Committee members
J. Saw (Chair, Interventional Cardiologist), Vancouver General Hospital, Vancouver, Canada; J-J. Blanc (Electrophysiologist), Brittany, France; P. Kanagaratnam (Electrophysiologist), St Mary’s Hospital, London, UK; W. Schillinger (Interventional Cardiologist), University Medical Center Göttingen, Göttingen, Germany; P. Wester (Stroke Physician), Umeå Stroke Center, Karolinska Institutet, Danderyds Hospital, Stockholm, Sweden.
Acknowledgements
There are several investigators and institutions that participated in the study but could not be included as authors on the manuscript. We would like to acknowledge those investigators and institutions for their participation and data contribution.
Australia: Epworth Hospital, Tony Walton; Fiona Stanley Hospital, Vincent Paul; Royal Adelaide Hospital, Glenn Young; Specialist Cardiology, Jason Sharp. Belgium: Hôpital Civil Marie Curie, Adel Aminian; UZ Gasthuisberg, Werner Budts. Chile: Hospital Clinico San Borja Arriaran, Daniel Aguirre. Denmark: Rigshospitalet, Lars Soendergaard. Finland: Helsinki University Central Hospital (HYKS), Juha Sinisalo; Tampere University Hospital, Saila Vikman. France: CHRU Lille, Didier Klug; CHU d’Amiens, Jean-Sylvain Hermida; CHU du Bocage, Luc Lorgis; Hôpital Haut Leveque, Jean-Benoit Thambo; Hôpital Henri Mondor, Emmanuel Teiger. Germany: CardioVaskulares Centrum St. Katharinen, Horst Sievert; Harzklinikum Dorothea Christiane Erxleben GmbH, Sven Fischer; Internistische Klinik Dr. Mueller, Thorsten Lewalter; Klinikum Frankfurt Hochst, Thomas Massa; Rems-Murr-Klinik Winnenden, Thomas Eul; Universitätsklinikum Ulm, Jochen Woehrle; Zentralklinik Bad Berka GmbH, Christoph Geller; Zentrum fur Herzgesundheit/Kardiologie am Alice Hospital, Kai Magnusson. Hong Kong: Prince of Wales Hospital, Anna Chan. Israel: Sheba Medical Center, Michael Glikson. Italy: Azienda Ospedaliera S. Anna e S. Sebastiano, Paolo Golino; Fondazione Toscana Gabriele Monasterio, Sergio Berti; Nuovo Ospedale Civile Sant-Agostino-Estense, Paolo Magnavacchi; Ospedale dell’Angelo, Francesco Caprioglio; Ospedale Luigi Sacco, Jacopo Oreglia. The Netherlands: Amsterdam Academic Medical Center (AMC), Robbert de Winter. Norway: Rikshospitalet, Ketil Lunde. Poland: Slaskie Centrum Chorob Serca, Zbigniew Kalarus; The Cardinal Stefan Wyszynski Institute of Cardiology, Marcin Demkow; Spain: Clinica Universidad de Navarra, Ignacio Garcia Bolao; Hospital Clinic I Provincial de Barcelona, Xavier Freixa; Hospital de la Santa Creu I Sant Pau, Dabit Arzamendi; Hospital Universitario Infanta Cristina, Jose Ramon Lopez Minguez; Hospital Universitario Son Espases, Armando Bethencourt; Hospital Universitario Virgen Macarena, Rafael Ruiz Salmeron. Sweden: Karolinska University Hospital Huddinge, Magnus Settergren; Sahlgrenska University Hospital - Gothenburg, Jacob Odenstedt. Switzerland: Basel University Hospital, Christian Sticherling. United Kingdom: Liverpool NHS Trust, Dhiraj Gupta; St. Thomas’ Hospital, Brian Clapp; University Hospital North Staffordshire, Robert Butler.
The authors would also like to thank Ashish Oza, Binh Ngo, Hong Zhao and Wim Stegink from Abbott for their help with the analysis and review of the manuscript.
Funding
The study was funded by St. Jude Medical/Abbott.
Conflict of interest statement
U. Landmesser reports personal fees from Abbott, Amgen, Sanofi, The Medicines Company, Bayer, and grants from Bayer, outside the submitted work. B. Schmidt reports personal fees from Abbott, during the conduct of the study, and personal fees from Abbott, and Boston Scientific, outside the submitted work. J.E. Nielsen-Kudsk reports grants from Abbott, during the conduct of the study, and is a proctor for Abbott. J-W. Park is a proctor for St. Jude Medical (now Abbott), Lifetech, OCCLUTECH and Cardia. G. Tarantini is a proctor for St. Jude Medical. I. Cruz-Gonzalez reports personal fees from St. Jude Medical, outside the submitted work, and is a proctor and consultant for St. Jude Medical. V. Geist is a proctor for St. Jude Medical (now Abbott). T. Zeus reports personal fees from St. Jude Medical, outside the submitted work. H. Omran reports personal fees from Bayer, Sanofi, Abbott, AstraZeneca, Medtronic. C. Piorkowski reports grants, personal fees and non-financial support from Abbott, during the conduct of the study, and grants and personal fees from Biosense, Medtronic, and Biotronik, outside the submitted work. J. Lund reports personal fees from St. Jude Medical during the conduct of the study, and personal fees from St. Jude Medical outside the submitted work. D. Hildick-Smith reports personal fees from St. Jude Medical, outside the submitted work. The other authors have no conflicts of interest to declare.
