
Abstract
Aims: Among antirestenotic compounds, sirolimus displays a superior safety profile compared to paclitaxel, but its pharmacokinetic properties make it a challenging therapeutic candidate for single-time delivery. Herein we evaluate the feasibility of delivery, long-term retention and vascular effects of sirolimus nanoparticles delivered through a novel porous angioplasty balloon in normal porcine arteries and in a swine model of in-stent restenosis (ISR).
Methods and results: Sirolimus nanoparticle formulation was delivered via porous balloon angioplasty to 753 coronary artery segments for pharmacokinetic studies and 26 segments for biological effect of sirolimus delivery in different clinical scenarios (de novo [n=8], ISR [n=6] and following stent implantation [n=12]). Sirolimus coronary artery concentrations were above the target therapeutic level of 1 ng/mg after 26 days, and were >100-fold higher in coronary artery treatment sites than in distal myocardium and remote tissues at all time points. At 28 days, reduction in percent stenosis in formulation-treated sites compared to balloon angioplasty treatment was noted in all three clinical scenarios, with the largest effect seen in the de novo study.
Conclusions: Local coronary delivery of sirolimus nanoparticles in the porcine model using a novel porous balloon delivery system achieved therapeutic long-term intra-arterial drug levels without significant systemic residual exposure.
Abbreviations
BMS: bare metal stent
DES: drug-eluting stent
ISR: in-stent restenosis
PCI: percutaneous coronary intervention
PK: pharmacokinetics
POBA: plain old balloon angioplasty
Introduction
Balloon-based delivery systems offer advantages over drug-eluting stents (DES) including: a) a therapeutic option for coronary lesions in which the stent-based approach is less desirable (e.g., small vessels), b) more drug delivered to the target area given the surface-area limitation of current stent technology, c) a reduction in the potential hypersensitivity response and inflammation related to the permanently implantable materials, and d) therapeutic options for non-coronary vessels for which DES therapy has been unsuccessful1-3. The ideal balloon-based drug delivery system requires the rapid, single-time, homogenous transfer of an antiproliferative agent to the arterial wall resulting in prolonged, site-specific uptake without washout or distal tissue exposure. As such, paclitaxel-coated balloons have emerged as a viable therapeutic alternative for patients undergoing percutaneous coronary intervention (PCI) for de novo, small vessel, and both bare metal stent (BMS) and DES ISR lesions2,4,5.
Due to its inherent pharmacokinetic features, paclitaxel has been the only drug used in the development of these technologies6. The use of sirolimus and its analogues in balloon-based delivery technologies has been limited by their intrinsic molecular instability once in solution and inability to achieve long-term tissue retention when delivered into local arterial tissue1,7. Sirolimus, however, remains an attractive option to prevent neointimal proliferation and subsequent vascular remodelling following balloon injury given its cytostatic mode of action and large safety margin compared to paclitaxel8.
Recent work done with limus-coated balloons includes a mixture of zotarolimus and iopromide in a balloon coating, used to treat porcine coronary arteries (followed with a BMS)9 or porcine femoral arteries10. Despite reductions in neointimal growth associated with the balloon coating, long-term sirolimus tissue levels were not demonstrated following a single balloon-based delivery11. Nanoparticle encapsulation technologies have recently emerged as an alternative method of formulating antiproliferative agents, and may overcome many of the barriers limiting the use of sirolimus12,13. By combining nanoparticle encapsulation with balloon angioplasty, a drug could be delivered to target tissues while maintaining favourable drug degradation and release profiles, e.g., phospholipid-coated sirolimus nanoparticles in a balloon coating were delivered to porcine coronary arteries (followed by BMS implantation) with tissue retention up to 14 days12. In this paper, we report on the pharmacokinetic and safety profiles of a novel sirolimus-eluting balloon device (Virtue™ Sirolimus-Eluting Balloon; Caliber Therapeutics, New Hope, PA, USA) using de novo and in-stent restenosis (ISR) porcine coronary artery models14,15.
Methods
DRUG-ELUTING BALLOON ANGIOPLASTY DILATATION SYSTEM
Standard percutaneous transluminal coronary angioplasty catheters (140 cm, rapid exchange) built with conventional, semi-compliant angioplasty balloons (diameter 2.5-4.0 mm; length 15-20 mm) are laser drilled with holes in uniform patterns (diameter 5-12 µm) around the cylindrical portion of each balloon. The resulting porous balloon catheters can thus be “primed” with a liquid formulation (driving ambient air out through the balloon pores), inflated with formulation, and pressurised to perform angioplasty and deliver formulation through the pores to the vessel wall simultaneously (Figure 1A). Despite the continuous flow of formulation during angioplasty, the compliance rating for the porous balloons is similar to normal angioplasty balloon catheters, with nominal size being reached at 10 atm. The number of holes per balloon is proportional to the balloon surface area. The balloon catheter is made up of the following sterile components: 1) sirolimus-eluting balloon catheter, 2) formulation reservoir, 3) lyophilised sirolimus formulation, and 4) high-pressure stopcock. The formulation reservoir is attached to the catheter, and the stopcock is located between it and the inflation device (indeflator), all via Luer fittings. Reconstituted formulation is introduced into the formulation reservoir and catheter via the side port of the stopcock.
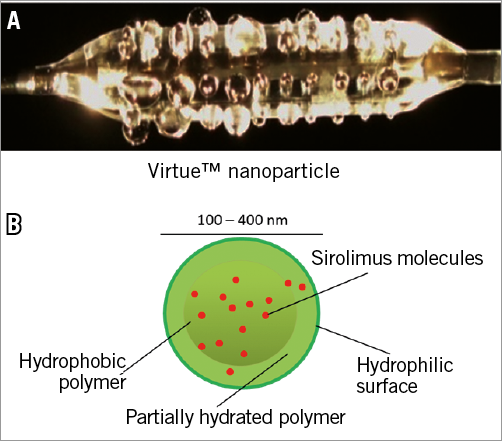
Figure 1. Caliber Virtue™ sirolimus-eluting angioplasty balloon and formulation. A) Image of expanded balloon eluting water. B) Schematic of nanoparticle containing sirolimus within the polymeric matrix; drug molecules migrate to surface prior to release.
IN VIVO SIROLIMUS DELIVERY PROCEDURE
Lyophilised sirolimus formulation is re-suspended in water or saline (sometimes containing contrast medium to enable visualisation of the expanded balloon via fluoroscopy), filtered, and loaded into the angioplasty balloon catheter via the stopcock between the formulation reservoir and indeflator, allowing trapped air to escape through the porous balloon. A direct fluid connection is made with the indeflator, which contains normal heparinised saline. A sufficient volume of formulation is loaded to eliminate delivery of diluted formulation. Once the balloon is positioned at the target site, delivery is commenced by turning the indeflator handle to inflate the balloon to 10 atm, which apposes the balloon to the vessel wall and initiates flow through the balloon pores. Angioplasty and drug delivery are continued by maintaining/increasing pressure to maintain flow, with an average final pressure of approximately 12 atm and average inflation time of 50 seconds to deliver a predetermined dose volume. Delivery times, pressures, balloon-artery expansion ratios, and delivery volumes were recorded for each treated segment. Delivered dose volumes were normalised to balloon surface area to yield dose density in µg/mm2.
NANO-ENCAPSULATED SIROLIMUS FORMULATION
Sirolimus is packaged, with biodegradable polyester-based polymers, in sub-micron nanoparticles that are stable in aqueous suspension (Figure 1B). The formulation is lyophilised with lyoprotectants in glass vials where it remains stable at room temperature. Sterility is achieved via either aseptic production techniques, or gamma or e-beam radiation. Vial contents are re-suspended in aqueous medium and filtered prior to use through syringe filters. Particle size and distribution measurements are performed with a Zetasizer Nano S (Malvern Instruments, Malvern, United Kingdom) with samples diluted 500x in 10 mM NaCl (25ºC, 120 second equilibration, dispersant=sample viscosity, automatic attenuation, normal resolution).
IN VITRO SIROLIMUS RELEASE ASSAY
Sirolimus is both insoluble and labile in aqueous solution, yet it remains suspended at high concentrations while nano-encapsulated. Thus, the release assay is most accurate measuring drug remaining within the nanoparticles, since eluted drug tends to degrade and/or precipitate. Formulation is re-suspended in water and dialysed against excess water or saline buffer at 37ºC. Sirolimus molecules elute from the nanoparticles into the aqueous buffer and through the dialysis tubing, where they degrade via hydrolysis or precipitate. At designated time points during incubation (one, 72 and 168 hours) dialysis containers are harvested and their sirolimus content quantified by high-pressure liquid chromatography. Data are expressed as percent remaining of the initial concentration (n=4 per time point).
IN VIVO PHARMACOKINETICS AND TISSUE EFFECT (HISTOLOGICAL) STUDIES
Studies were performed at the CRF-Skirball Center for Innovation. Domestic farm swine (30-50 kg; female or castrated male) were treated in accordance with the regulations outlined in the United States Department of Agriculture (USDA) Animal Welfare Act (9 CFR, Parts 1, 2 and 3) and the conditions specified in The Guide for Care and Use of Laboratory Animals (ILAR publication, 2011, National Academy Press). The test facility is accredited by the Association for the Assessment and Accreditation of Laboratory Animal Care, International (AAALAC) and registered with the USDA. Study protocols (not GLP compliant) were approved by the Institutional Animal Care and Use Committee. Animals were pre-treated with 325 mg of aspirin and 150 mg of clopidogrel bisulphate administered one day before the procedure. Following anaesthesia induction with tiletamine/zolazepam, xylazine and glycopyrrolate, they were intubated and general anaesthesia was maintained with inhaled isofluorane (1-3%). A vascular access sheath was placed in the common carotid artery by cutdown. Before catheterisation, heparin was injected to maintain an activated clotting time of >250 seconds. Access to coronary arteries was via a 6 Fr guiding catheter. Vessel size and delivery locations were determined by quantitative coronary angiography. Exact locations of treatment sites were identified based on a combination of angiograms and anatomic landmarks, sometimes including stent placement. Sirolimus formulation was delivered to 779 coronary vessel segments in 130 domestic swine. A total of 753 coronary artery segments were used for the PK study (Figure 2A), either in de novo (706 segments) or in the ISR setting (47 segments). In addition, 26 coronary artery segments were used to evaluate the biological effect of sirolimus delivery in different clinical scenarios (de novo [n=8], ISR [n=6] and following BMS implantation [n=12]), and their treated vessels were prepared and analysed for histomorphometry (Figure 2B). All deliveries were performed using an average balloon-to-artery ratio of 1.3.
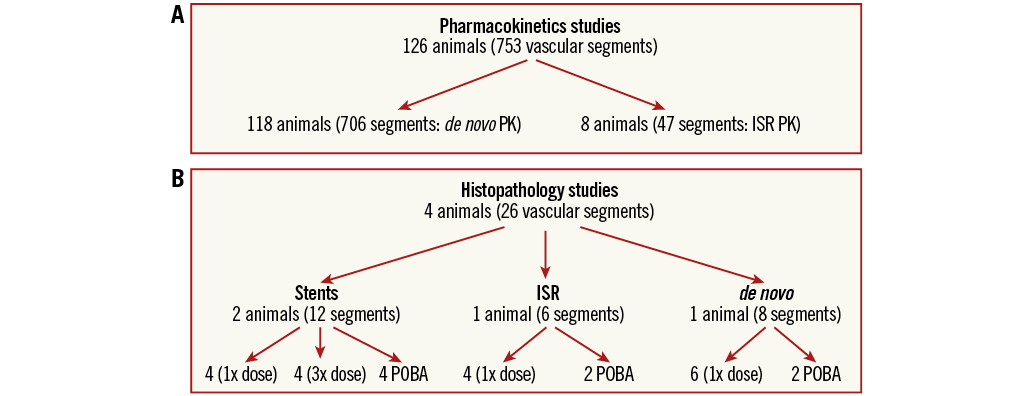
Figure 2. Pharmacokinetic and histopathology study designs for analysis of coronary treatment sites following deployment of the indicated device. A) Pharmacokinetics studies. B) Histopathology studies.
For PK studies, treated arteries (and other tissues) were excised, frozen on dry ice, and stored at −80°C. For histological studies, treated hearts were explanted, flushed with Lactated Ringer’s solution, perfused with 1 litre of 10% neutral buffered formalin (NBF), and immersed in a container of 10% NBF for a minimum of 24 hours prior to being sent for histological analysis. The ISR model consisted of balloon injury (1.3 balloon:artery ratio) followed by stent implantation (1.2 stent:artery). After 28±7 days, in-stent restenosis was clearly observed by angiography and histology15. Sirolimus-eluting balloon treatments targeted the same diameter as the stent placement (20% overstretch) for consistent deliveries with relation to the original vessel reference vessel diameter (RVD). The delivery parameters (time and pressure) were consistent with this model. Tissue samples were dissected away from stents prior to analysis.
HIGH-PRESSURE LIQUID CHROMATOGRAPHY SIROLIMUS ASSAY
Samples from formulation vials, in vitro release assays and blood samples were vortexed for 30 seconds in acetonitrile. Tissue samples were weighed, cut into small pieces, incubated in acetonitrile for 30 minutes, and homogenised for 30 seconds using a high-speed tissue homogeniser. All samples were centrifuged for 10 minutes at 10k rpm in a table-top micro-centrifuge to remove debris, loaded onto an Agilent 1100 HPLC (Agilent, Santa Clara, CA, USA), and run with the same programme (C18 column; mobile phase, 85% acetonitrile/15% water; flow rate, 1.0 mL/min; temperature, 35ºC; detector, UV 278 nm; sample volume, 20 µL; run time, 12 minutes). Sirolimus concentration was assessed in relation to a standard curve run from newly dissolved sirolimus.
SYSTEMIC PHARMACOKINETICS
PK animals received an average of ~6 coronary deliveries per procedure. These multiple deliveries provided an average total dose of 29.66±13.52 mg sirolimus per animal, or 0.76 mg/kg. By contrast, the human clinical dose (from a single deployment with the largest device) would be 0.049 mg/kg, or about 15-fold lower. Animals included in the analysis of distant organs received an average dose of 20.4 mg per animal, or about 10 times the largest clinical dose. Blood samples were taken from seven animals, timed following the final treatment. Total delivered sirolimus dose ranged from 4.11 mg to 23.65 mg (12.9±7.2 mg, mean±SD), with average dose approximately 6.8 times the clinical dose from the largest device.
Results
IN VITRO CHARACTERISATION OF NANOPARTICLE SIROLIMUS FORMULATION
During formulation development, more than 670 sirolimus nanoparticle lots were prepared, all with polymeric components uniformly combined with sirolimus (average particle diameter range 114.9 nm to 391.9 nm). Two in vitro characteristics monitored were drug content stability and elution profile. Stability of lyophilised formulations was tested after prolonged storage at room temperature, comparing sirolimus levels to those measured immediately after lyophilisation. The nanoparticle-encapsulated formulation demonstrates stable sirolimus content levels during their time in storage (Figure 3A). In vitro sirolimus elution calculated upon the suspension of the nanoparticles in an aqueous medium is shown in Figure 3B, illustrating that the formulation tested soon after lyophilisation (t0) demonstrates slow release of sirolimus, whereby approximately 50% of the drug remains intact within the nanoparticles after one week in aqueous suspension at 37ºC. Moreover, this characteristic does not change when measured in formulation subjected to room temperature storage for six, 13, or 26 weeks prior to re-suspension.

Figure 3. In vitro properties of the sirolimus nanoparticle formulation. A) Drug content stability of Virtue therapeutic formulation in vitro. Sealed sterile vials containing the lyophilised formulation were stored for given times at room temperature (RT 20-25ºC; ambient humidity, dark); vials were reconstituted and analysed for sirolimus content; % remaining was calculated in comparison with time zero; mean±SEM, sample sizes range from five (six, 13, and 26 weeks) to 19 (post-sterile baseline). B) Functional stability of therapeutic formulation. Sealed sterile vials of formulation were reconstituted after given storage times at room temperature (20-25ºC) and analysed with the sirolimus release assay; expressed as percent remaining of the zero time point. Mean±SEM.
TARGET TISSUE AND SYSTEMIC PHARMACOKINETICS
Target tissue PK studies involved 126 animals in which 753 coronary sites were treated. A decrease in sirolimus tissue concentration was evident with increased survival time (Figure 4A). Coronary artery sirolimus concentration achieved levels above ~30 ng/mg at seven days and remained substantially higher than the therapeutic target of 1 ng/mg at later time points (~12 ng/mg at 26 days). The analysis of sirolimus concentration in distant organs is illustrated in Figure 4B. Dissected myocardium demonstrated about 100-fold lower acute levels than in coronary tissue, with significant reductions after one day, and virtually undetectable levels after one week. A profile of distant organs (kidney, liver, and lung) receiving systemic exposure to formulation revealed several fold lower sirolimus levels than myocardium, with the lung displaying the highest acute levels among the three organs. Sirolimus levels were undetectable after four days in the kidney and liver, and after six days in the lung.
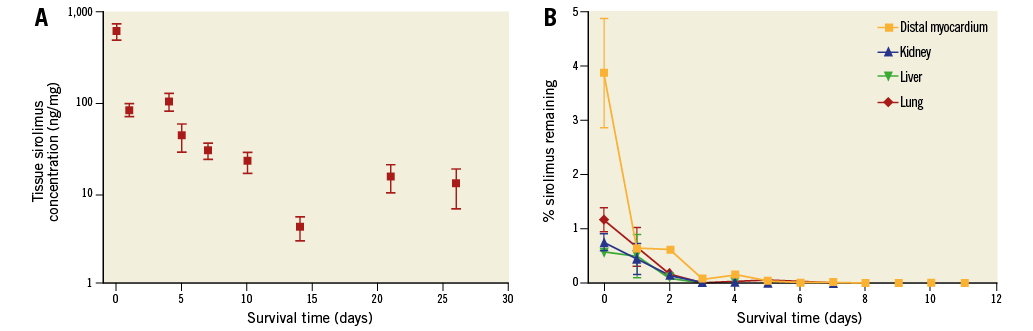
Figure 4. In vivo pharmacokinetics of the sirolimus nanoparticle formulation. A) Sirolimus tissue levels in porcine coronary arteries. Treated coronary artery segments harvested, weighed and analysed for sirolimus content, expressed as ng sirolimus per mg wet tissue weight; sample sizes range from 15 (26 days) to 247 (seven days). B) Non-target porcine tissue levels. Distal myocardium and non-target organ samples were harvested, weighed and analysed for sirolimus content, expressed as ng sirolimus per mg wet tissue weight.
Sirolimus was detected in blood at one hour (409±78 ng/ml) and rapidly declined at 24 hours (66±24 ng/ml), being undetectable beyond 96 hours. The maximum tissue concentration (Cmax) in these animals was 416±77 ng/mL and occurred at a Tmax of 11±8 min following the final drug administration.
EVALUATION OF BIOLOGICAL TISSUE EFFECT
The biological effect of nanoparticle sirolimus delivery was performed simulating three different clinical scenarios. A variety of morphometric and safety-related (histological) parameters are summarised from analysis of both coronary arteries and downstream myocardial tissue (Table 1, Figure 5). A slight reduction in percent stenosis in formulation-treated sites compared to plain old balloon angioplasty (POBA) treatment was noted in all three studies. The reduction was modest and associated with 3x dosing in the same-day stenting of the treatment sites (Figure 5A); the reduction was also modest in the ISR study (Figure 5B), but more dramatic in the de novo study (Figure 5C). The same pattern of formulation effect was seen in neointimal thickness: reductions associated with the 1x dose in the ISR and de novo studies, and a modest reduction in the 3x group in the same-day stenting study. A noteworthy increase in mineralisation score associated with formulation treatment was also noted. The ISR study showed the greatest formulation-related reduction in inflammation score, whereas the same-day stent study showed less inflammation in the POBA group than in the 3x dosing group, and neither group showed any indication of inflammation in the de novo study. Injury scores were generally low, especially in the stented and de novo studies. The distal myocardial bed was analysed in both ISR and de novo studies, with no infarction or changes ascribable to treatment reported for either.
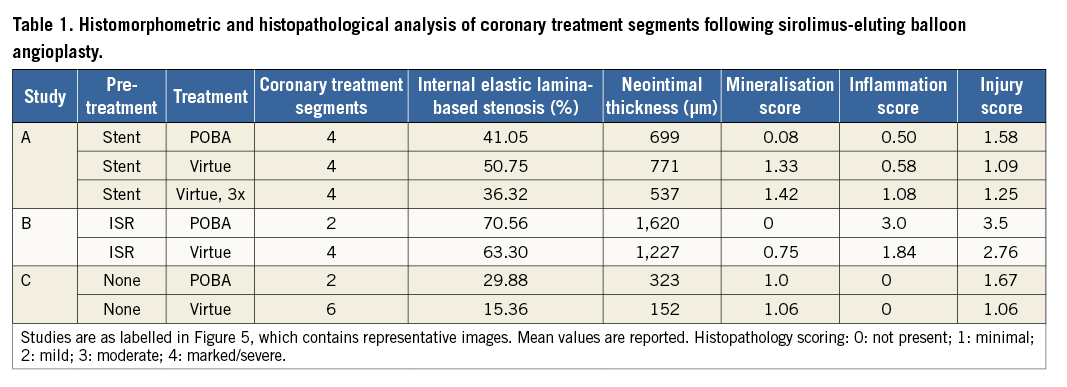
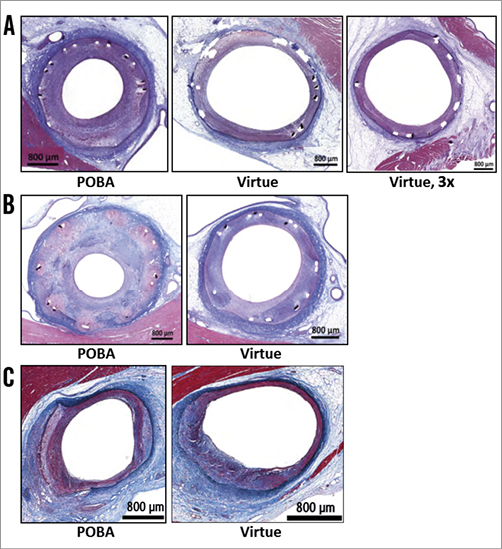
Figure 5. Representative histologic images following sirolimus-eluting angioplasty balloon treatment. Porcine vessels were harvested for analysis 28 days after angioplasty or drug delivery following BMS implantation (A), drug delivery in ISR setting (28 days) (B), and de novo vessels (C). Day 0 treatment was with plain old balloon angioplasty (POBA) or sirolimus-eluting angioplasty balloon at nominal 1x dose (Virtue) or 3x dose (Virtue, 3x).
Discussion
Drug-coated balloons provide a therapeutic alternative for patients undergoing PCI by achieving prolonged and site-specific uptake of antiproliferative agents to the coronary arterial wall1. Paclitaxel has been studied extensively and has become the drug of choice for use in balloon-based delivery systems6. Sirolimus offers superior clinical effectiveness given its safety profile, mode of action, and capacity for reduced vascular remodelling following vessel injury8. By combining a novel nanoparticle encapsulation technology with an angioplasty balloon dilatation system, we have demonstrated the first evidence of prolonged sirolimus tissue levels following a single balloon-based delivery.
Studies have been performed in the healthy porcine coronary artery model, both to gain insight into the pharmacokinetics and safety of the approach, and to test the device in a manner similar to its intended use in the clinical setting. While this model is not ideal for demonstrating clinical efficacy, its physical and biological similarities to humans support its potential as a surrogate for clinical efficacy in the context of drug uptake and retention11,15. The data presented here indicate a tissue retention profile consistent with efficacious treatment of restenosis. This is in contrast to arteries treated with sirolimus (or analogue) balloon-based coatings, that have not shown long-term drug retention1,9,10,12,13. Drug levels in downstream myocardium (from treated vessels), distant organs (kidney, liver and lung), and whole blood indicate that sirolimus not retained in target tissue is short-lived and unlikely to pose safety concerns. Histological analysis of treated arteries and downstream myocardium shows no indication of pathology or toxicity resulting from the treatments. Notably, the distal myocardial bed, despite exposure to the nano-encapsulated formulation, was found to display no infarction or other changes ascribable to treatment. Moreover, the systemic PK measurements in this study are from animals which received much higher sirolimus doses than patients will receive in the clinical setting, suggesting even lower systemic exposure and a shorter residence time of drug in patients. Finally, morphometric analysis of treated artery segments suggests that the device has the potential to treat restenosis in both stented and non-stented vessels.
The presented technology was designed to perform balloon angioplasty and deliver a reconstituted nanoparticle suspension containing sirolimus to the coronary artery lesion simultaneously. The many variables involved in development of this technology include balloon pore configuration, dose volume, delivery pressure, duration of delivery, and balloon-to-artery ratio. Although in vitro tests are performed whenever possible, only the in vivo model combines all of the relevant variables to permit assessment of device performance as measured by drug uptake and persistence in the coronary artery tissue. The large number of treated vessels in these studies reflects the demands of testing several variables. The porcine coronary artery model was chosen, as recommended, for its similarity to humans15, with the additional advantage of its capacity to accommodate many treatments per animal. Most testing of this device has been done on healthy arteries, where the average balloon-to-artery ratio during angioplasty of 1.3 mimics the dynamics of treating a diseased vessel in the clinical setting, e.g., a diseased vessel with 25% diameter stenosis expanded to RVD would experience a similar 1.3 balloon-to-lumen ratio.
Measurement of target tissue drug levels (PK analysis) serves as a surrogate measure of efficacy based on the known effects of sirolimus in similar model systems such as drug-eluting stents in porcine coronary arteries. This is a vital metric in the development of a device for which clinical efficacy endpoints are logistically and statistically difficult to demonstrate in the preclinical setting, where animal models display differences in health status, age, and pharmacokinetic response to therapeutics from the human patient population. The expectation is that therapeutic levels of sirolimus must persist in the target tissue for a critical period of three to four weeks following angioplasty to achieve antirestenotic efficacy. Based on sirolimus-eluting stent (analogous to the CYPHER® stent; Cordis Corporation, Fremont, CA, USA) studies, a sirolimus tissue level of 1 ng/mg after four weeks is sufficient to inhibit stenosis11,16. More recently, a stent coated with a sirolimus analogue, everolimus, was found to leave 0.69±0.30 ng/mg drug in the target tissue after four weeks17. Other porcine studies have demonstrated sirolimus concentrations in the 2-5 ng/mg range 30 days following stent implantation18. Additionally, histological analysis of treated artery segments provides a measure of efficacy and safety based on the reduction of neointimal growth (efficacy) and absence of significant inflammation/injury (safety) four weeks after treatment19. The limited number of histological studies presented herein offers encouraging indications in this regard.
Measurement of non-target tissue PK addresses an important aspect of device safety, since liquid formulation that is not absorbed by the target coronary arterial tissue flows downstream into the general circulation, thus exposing distant organs while in the process of being eliminated. The ability of the device to deliver efficacious dosing to the target tissue, while not flooding the circulation or downstream organs with excessive amounts of sirolimus, speaks to the likelihood that the therapeutic benefit of the device will come with minimal risk of off-target side effects. The measured sirolimus concentrations in both treatment and non-treatment tissues compare favourably with recommended sirolimus concentrations in approved therapies, e.g., organ rejection, where a 6 mg loading dose followed by daily oral doses of 2 mg/day are intended to achieve blood levels of 8-20 ng/mL, although loading doses of 15 mg followed by daily oral doses of 5 mg have also been found to be safe20. It is apparent that, after clinical treatment with the current device, blood concentrations of sirolimus would be within the 8-20 ng/mL range after one day. Clinical studies of oral sirolimus for restenosis (five randomised trials) have demonstrated effectiveness compared with BMS, but not DES, alone; loading doses (0-24 mg range) were followed with 2-3 mg/day for seven to 30 days, with average cumulative doses of 60 mg21.
Finally, several aspects of device safety have been addressed by histology studies. No safety concerns were noted in any of the studies, and histomorphometric analysis suggested a biological effect on neointimal proliferation comparable to the one produced by metallic drug-eluting stents. Notably, an increase in mineralisation score (commonly used to indicate the presence of a polymer) was associated with formulation treatment in both ISR and same-day stents. Additionally, the POBA and formulation groups showed near-equal mineralisation in the de novo study. Finally, injury scores were generally low, especially in the stented and de novo studies, and were the highest in the POBA group, consistent with the idea that angioplasty-related injury is responsive to treatment with sirolimus.
In summary, in these studies we have demonstrated the feasibility, safety and potential for efficacy of a novel approach for delivering a long-acting nano-encapsulated sirolimus formulation to target coronary arteries via dedicated porous angioplasty balloon technology. Further studies are needed to test the effectiveness of the device in humans and to compare it to existing paclitaxel-coated balloon technologies.
| Impact on daily practice All currently available drug-coated angioplasty balloons deliver paclitaxel to prevent restenosis. Sirolimus, a proven antirestenotic drug with a preferred toxicity profile, has not demonstrated long-term elution or effect when delivered via a balloon coating. The present study shows that a sirolimus nanoparticle formulation delivered via porous angioplasty balloon provides long-term sirolimus retention in porcine coronary arteries. This would provide a significant therapeutic alternative for interventional cardiologists in cases where stenting and/or the use of the cytotoxic drug paclitaxel are not desired. |
Conflict of interest statement
The present study was sponsored by Caliber Therapeutics, Inc. (New Hope, PA, USA). W. Baumbach, B. Bingham, and Y.F. Keng are full-time employees, and G. Stone and M. Leon are minor equity holders of Caliber Therapeutics, Inc. The other authors have no conflicts of interest to declare.
