
Dual antiplatelet therapy with aspirin and an inhibitor of the platelet P2Y12 receptor (clopidogrel, prasugrel, and ticagrelor) represents the cornerstone of treatment for patients undergoing percutaneous coronary intervention (PCI), including those presenting with ST-segment elevation myocardial infarction (STEMI) undergoing primary PCI (PPCI)1. In patients undergoing PPCI, pharmacokinetic (PK) and pharmacodynamic (PD) studies have consistently shown a delay in the antiplatelet effects of oral P2Y12 receptor inhibitors in the early hours after STEMI presentation, including with the more potent agents prasugrel and ticagrelor even when used at high dosing regimens, due to impaired gastrointestinal absorption2,3. Crushing tablets has been shown to improve the PK/PD profiles of prasugrel and ticagrelor, providing faster onset of platelet inhibition after PPCI4-6. However, these studies have been conducted primarily in non-comatose patients who are able to swallow and who do not need a nasogastric tube (NGT).
Patients undergoing PPCI following a cardiac arrest require their antiplatelet medication to be administered through a NGT, typically in a crushed formulation. However, there are limited data on the PK/PD profile of oral P2Y12 receptor inhibitors in this particular setting. The NGT, per se, may affect drug bioavailability due to the fact that the medication may adhere to the inner coating of the tube. Moreover, many of these patients are also in cardiogenic shock, a status which compared with patients not in cardiogenic shock is associated with impaired gastrointestinal absorption due to vasoconstriction as well as the use of opiates for sedation2,3. Finally, many patients post out-of-hospital cardiac arrest are treated with therapeutic hypothermia (TH), which is also known to reduce platelet inhibition induced by oral P2Y12 receptor inhibitors7-10. Most of the currently available data in patients treated with TH after cardiac arrest derive from PD studies assessing the effects of clopidogrel, which have been shown to be markedly reduced7,8. On the other hand, the PD effects of prasugrel and ticagrelor (administered as crushed tablets via an NGT) have been shown to be affected to a lesser extent than clopidogrel and to provide more prompt and effective platelet inhibition without significant increases in rates of high on-treatment platelet reactivity (HPR), a well-established marker of thrombotic complications9,10. These findings may be attributed to the fact that TH is associated with a decrease in hepatic cytochrome P450 activity, which is a key determinant of clopidogrel metabolism9.
In this issue of EuroIntervention, Ratcovich et al report the results of the TICOMA (Ticagrelor for the Comatose) study, which investigated the PD and PK effects of ticagrelor in comatose patients undergoing PPCI11. In particular, in this prospective observational single-centre study, 44 patients resuscitated but comatose after out-of-hospital cardiac arrest and treated with PPCI were enrolled; of these, 41 were also treated with TH. A crushed 180 mg loading dose of ticagrelor, followed by a 90 mg bid maintenance dose, dissolved in 50 ml of purified water was administered through an NGT immediately after PPCI. Blood samples for PD and PK assessment were performed before the ticagrelor loading dose and two, four, six, eight, 12, and 24 hours after, and then daily for an additional five days. Platelet reactivity was measured by VerifyNow® (Accriva, San Diego, CA, USA) and Multiplate® analyser (Cobas/Roche Diagnostics International AG, Rotkreuz, Switzerland), and PK assessments included the measurement of plasma levels of ticagrelor and its major active metabolite (AR-C124910XX). The authors found that platelet reactivity decreased over the study time course following ticagrelor loading dose administration, and HPR rates at 12 hours (primary endpoint) were 12% and 7% by VerifyNow and Multiplate, respectively. The median time to reach adequate platelet inhibition was three and four hours with VerifyNow and Multiplate, respectively. Peak plasma concentrations of ticagrelor and AR-C124910XX were found to be lower compared to those from studies conducted in non-comatose patients3,11.
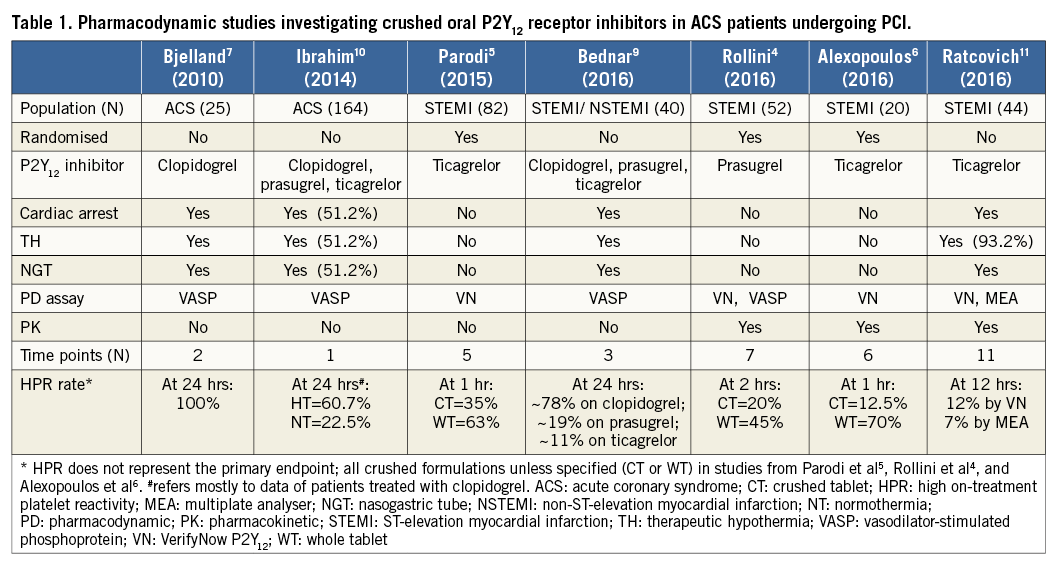
The authors must be congratulated for this study. The major strengths of this investigation are that, for the first time, both the PD and PK profiles of crushed ticagrelor in a real-world population of comatose patients undergoing PPCI have been studied, with assessments being conducted at multiple time points, and platelet reactivity corroborated by two different and well-established assays (Table 1). However, some limitations also need to be considered. As acknowledged by the authors, there were no PK/PD assessments performed between baseline and two hours. The lack of data during this time frame is critical given that that is when platelet inhibition by ticagrelor is known to be mostly impaired in non-comatose PPCI patients. In line with this, the choice of 12 hours as the timing for the primary endpoint is questionable, as the thrombotic risk following PPCI is known to be highest in the very early hours after loading dose administration, which also reflects the window of vulnerability associated with impaired platelet inhibitory effects. Although the authors state that the time to reach adequate platelet inhibition was approximately three hours, there was high inter-patient variability, with many patients probably experiencing HPR in the first eight hours. In addition, the wide window allowed for the timing of blood draw (±1 hour) at each time point might have added significant variability to the study results. The authors also did not enrol non-comatose patients receiving ticagrelor through an NGT to use as a control group, which would have allowed more accurate comparisons.
In conclusion, the TICOMA study demonstrates that crushed ticagrelor administered through an NGT can provide acceptable levels of platelet inhibition in comatose patients treated with PPCI. However, these levels are achieved only after several hours of drug administration. Although studies investigating the association between the use of TH and acute stent thrombosis have yielded inconsistent findings, these studies were heterogeneous in the choice of antithrombotic medications and were mostly underpowered to detect difference in stent thrombosis rates12. Although it may be difficult to find a causal relationship between a rare event such as acute stent thrombosis and TH, impaired platelet inhibition (i.e., HPR) is known to be associated with thrombotic complications and, intuitively, having better platelet inhibition is more desirable in high-risk patients undergoing coronary stenting1. This underscores the need for the use of intravenous agents, such as the P2Y12 receptor inhibitor cangrelor and the glycoprotein IIb/IIIa inhibitors, which are not affected by TH or impaired gastrointestinal absorption, and which thus represent attractive options to provide immediate and potent platelet inhibition in these patients during their period of highest vulnerability (i.e., the early hours after oral antiplatelet drug administration)13. Indeed, further studies are warranted to explore the clinical effects of these strategies.
Conflict of interest statement
D. Angiolillo has received consulting fees or honoraria from Sanofi, Eli Lilly, Daiichi-Sankyo, The Medicines Company, AstraZeneca, Merck, Abbott Vascular, Amgen, Bayer, Pfizer, Chiesi, Biosensors, and PLx Pharma. He has participated in review activities from CeloNova, Johnson & Johnson, and St. Jude Medical. He has also received institutional payments for grants received from Glaxo-Smith-Kline, Eli Lilly, Daiichi-Sankyo, The Medicines Company, AstraZeneca, Janssen Pharmaceuticals, Inc., Osprey Medical, Inc., Novartis, CSL Behring, and Gilead. The other authors have no conflicts of interest to declare.
