
Abstract
Aims: The Absorb bioresorbable vascular scaffold (Absorb BVS) provides similar clinical outcomes compared with a durable polymer-based everolimus-eluting metallic stent (EES) in stable coronary artery disease patients. ST-elevation myocardial infarction (STEMI) lesions have been associated with delayed arterial healing and impaired stent-related outcomes. The purpose of the present study is to compare directly the arterial healing response, angiographic efficacy and clinical outcomes between the Absorb BVS and metallic EES.
Methods and results: A total of 191 patients with acute STEMI were randomly allocated to treatment with the Absorb BVS or a metallic EES 1:1. The primary endpoint is the neointimal healing (NIH) score, which is calculated based on a score taking into consideration the presence of uncovered and malapposed stent struts, intraluminal filling defects and excessive neointimal proliferation, as detected by optical frequency domain imaging (OFDI) six months after the index procedure. The study will provide 90% power to show non-inferiority of the Absorb BVS compared with the EES.
Conclusions: This will be the first randomised study investigating the arterial healing response following implantation of the Absorb BVS compared with the EES. The healing response assessed by a novel NIH score in conjunction with results on angiographic efficacy parameters and device-oriented events will elucidate disease-specific applications of bioresorbable scaffolds.
Introduction
The Absorb everolimus-eluting bioresorbable vascular scaffold (Absorb BVS; Abbott Vascular, Santa Clara, CA, USA) was designed to degrade predictably within three years after implantation1. The scaffold has been assessed predominantly in patients with stable coronary artery disease2,3. Recent evidence from a randomised trial (ABSORB II)4 indicates similar clinical performance compared with metallic everolimus-eluting stents (EES) throughout one year of follow-up5. Bioresorbable scaffolds provide intriguing features including restoration of vasomotor function, late lumen gain owing to positive remodelling, plaque shielding properties and uncaging of previously stented segments, which may provide clinically important long-term benefits among patients undergoing coronary revascularisation6.
Potential of bioresorbable scaffolds in STEMI
Acute ST-elevation myocardial infarction (STEMI) represents a clinical entity which may be particularly suited for the use of bioresorbable scaffolds. In the vast majority of cases, STEMI lesions feature a large necrotic core with superimposed thrombus, which may prolong the arterial healing process and increase the inflammatory response to metallic drug-eluting stents (DES), as evidenced by post-mortem pathology analyses7. Along the same lines, optical frequency domain imaging (OFDI) studies have revealed a higher frequency of uncovered and malapposed stent struts in the lesions of STEMI compared with stable CAD patients during mid8 and long-term follow-up9. Notwithstanding this, malapposed struts may not only be the consequence of impaired healing and persistent inflammation, but may also occur during the procedure owing to inadequate stent apposition, or following the procedure due to thrombus resolution, a phenomenon more frequently observed in the setting of primary PCI. These aspects may be summarised as an increased risk of adverse arterial healing in the setting of primary PCI.
Lesions underlying STEMI are, in around two thirds of cases, non-calcified and located in the proximal segments of the epicardial vessels. These lesions are ideally suited for bioresorbable scaffolds owing to the large vessel diameter and the possibility of full scaffold expansion. The expected attenuation of late stent failures may be particularly effective in STEMI patients due to the proximal localisation of STEMI culprit lesions with a large myocardial area at risk. Moreover, patients with STEMI are on average five years younger than patients with stable CAD, increasing the long-term benefit of bioresorbable scaffolds in the absence of a permanent implant. Normalisation of vasomotion and compensatory remodelling have been reported following Absorb BVS implantation, suggesting the restoration of a normal vessel physiology after complete biodegradation of the scaffold struts10.
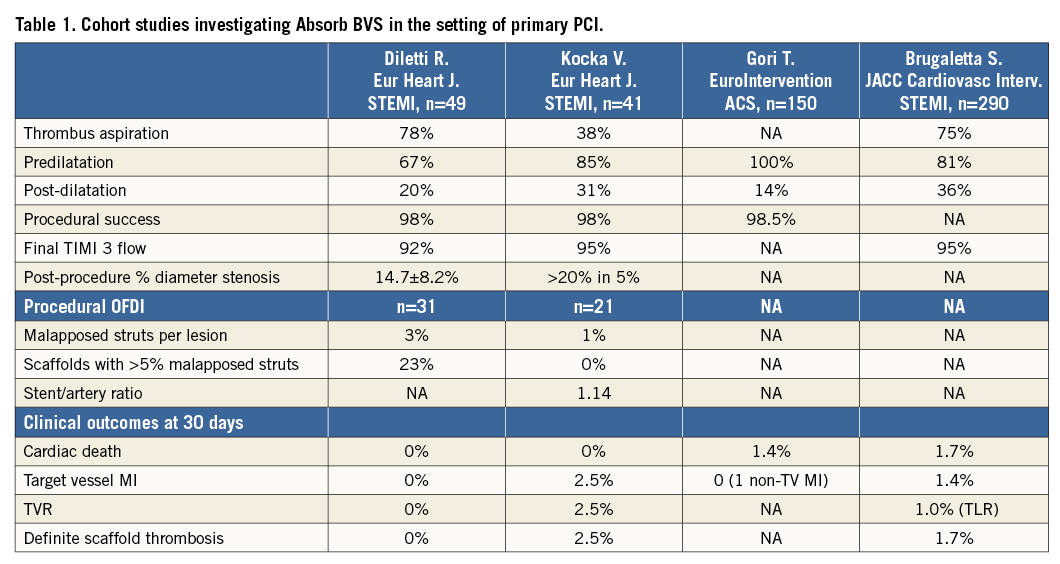
Available evidence in STEMI
New-generation DES stents (the biolimus-eluting BioMatrix™; Biosensors, Singapore, and everolimus-eluting XIENCE™; Abbott Vascular) have been shown to reduce cardiovascular events in STEMI patients, including the risk of target lesion revascularisation (TLR), myocardial infarction (MI) and stent thrombosis, as compared with metallic bare metal stents in large randomised trials11,12. The use of bioresorbable scaffolds in STEMI patients is limited to non-randomised studies with fewer than 500 patients reported to date13-15. Two observational studies included 41 and 49 patients, respectively, and employed OFDI at baseline and follow-up in a subgroup of patients (Table 1). Device implantation was successful in more than 98% of patients, with a TIMI 3 flow achieved in more than 92% of patients. Clinical event rates were low at 30 days, with only one case of scaffold stent thrombosis during follow-up. In addition, OFDI confirmed patent scaffolds with a low frequency of malapposed scaffold struts (<3%) in both studies. The largest observational STEMI registry enrolled 290 patients treated with the Absorb BVS scaffold, and compared clinical outcomes in a propensity score-matched analysis with everolimus-eluting XIENCE and bare metal stent-treated patients. At one year, the device-oriented composite endpoint cardiac death, target vessel MI and TLR did not differ between groups, although, numerically, definite and probable stent thrombosis occurred more frequently in BVS-treated patients16. To date, there is no randomised trial comparing the Absorb BVS scaffold with the newer-generation DES in terms of angiographic or clinical outcomes.
This study of arterial healing in response to the implantation of the Absorb BVS compared with a metallic EES will provide important insights to guide our understanding of scaffold implantation in the setting of primary PCI and may provide the basis for larger clinical endpoint trials. While observational data on the feasibility and procedural success of bioresorbable scaffold implantation in the setting of primary PCI exist, no randomised study has addressed this question so far. Although not powered for the comparative evaluation of cardiovascular outcomes, this study is anticipated to provide hypothesis-generating insights into the frequency of efficacy and safety parameters.
Methods
STUDY DESIGN
ABSORB TROFI II is a randomised, single-blind study comparing the Absorb BVS everolimus-eluting scaffold (Absorb BVS) with a metallic EES (1:1 allocation) (XIENCE PRIME; Abbott Vascular) in patients presenting with STEMI, and quantifies arterial healing six months after scaffold/stent implantation (Figure 1). The primary hypothesis of the ABSORB TROFI II study is that the arterial healing response of the Absorb BVS is non-inferior compared with the durable polymer EES, as assessed by a dedicated neointimal healing score using OFDI six months after the index procedure (Figure 2).
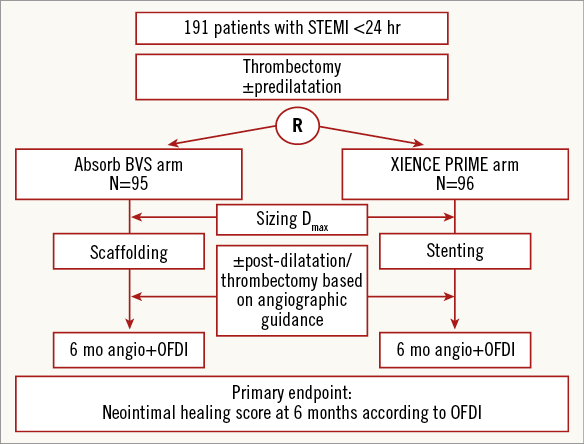
Figure 1. Study flow chart.
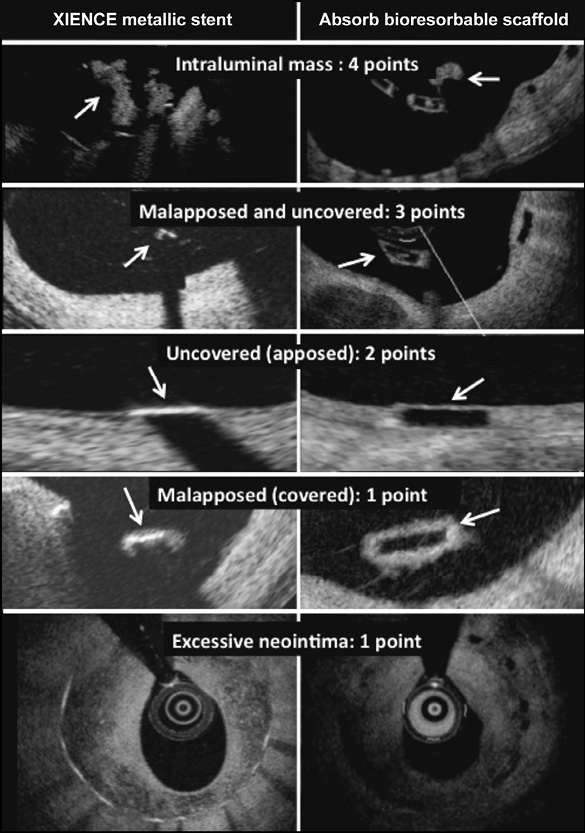
Figure 2. Illustration of the neointimal healing score. Arrows point to the intraluminal mass and to the uncovered and malapposed scaffold/stent struts.
Patients are blinded to the implanted stent type until the end of the study at three years. OFDI will be used to determine the neointimal healing score, which consists of scaffold/stent strut-related adverse healing characteristics (stent strut malapposition, uncovered struts and intraluminal masses). Randomisation is performed by dedicated web-based software after adequate lesion preparation using thrombectomy (and predilatation, if required).
PATIENT POPULATION
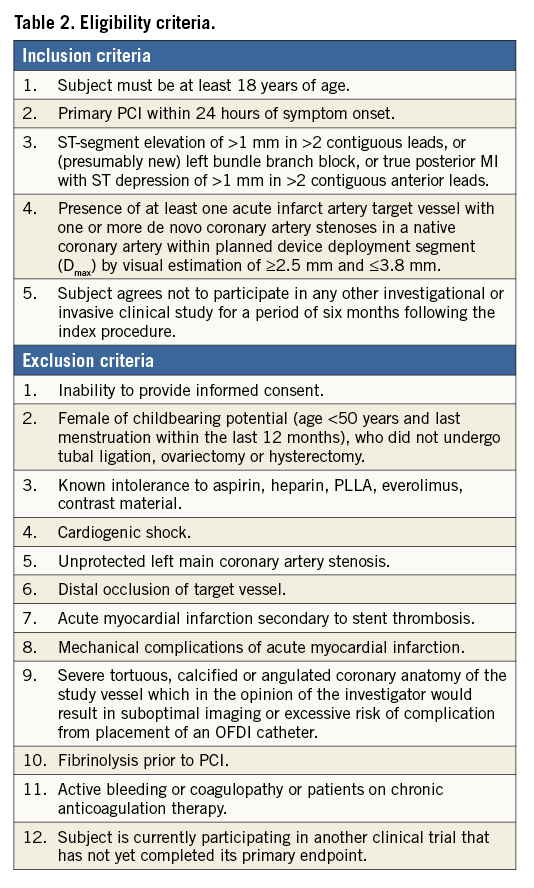
Patients presenting with STEMI who undergo primary PCI are eligible in the case of appropriate vessel size (according to maximal proximal and distal reference diameter), between 2.25 and 3.80 mm, and following adequate lesion preparation. Exclusion criteria include cardiogenic shock, severe tortuosity or calcification, and inadequate vessel size (<2.25 mm or >3.80 mm). Detailed inclusion and exclusion criteria are summarised in Table 2. Patients will be included at eight international sites located in Denmark, The Netherlands, Spain, and Switzerland (Table 3).
DEVICES
The Absorb BVS scaffold is available in diameters of 2.5, 3.0, and 3.5 mm and in lengths of 8, 12, 18, and 28 mm. It is recommended to use similar sizes for the EES, XIENCE Xpedition®.
PROCEDURE
Manual thrombus aspiration is mandatory in all culprit lesions and multiple thrombectomy passages will be performed (at least two) to minimise the angiographically visible thrombus burden.
Predilatation is at the discretion of the operator. If predilatation is performed, the balloon should be selected according to the reference vessel diameter (1:1) or somewhat smaller. The use of a non-compliant balloon is recommended in case of a 1:1 sizing. The full expansion of the predilatation balloon has to be confirmed, and in case of a residual stenosis of >40% the patient cannot be randomised. Randomisation can only be performed after adequate lesion expansion and establishment of at least TIMI 2 flow after thrombus aspiration and/or predilatation. Written informed consent must be obtained from all patients prior to randomisation.
Scaffold/stent sizing is based on the distal and proximal maximal vessel diameter 5-10 mm distal and proximal to the boundaries of the lesion defined by QCA. The maximal proximal and distal lumen diameter should be between 2.25 and 3.8 mm. A 3.5 mm scaffold/stent should be used in case of a maximal distal and proximal lumen diameter between 3.0 and 3.8 mm, a 3.0 mm scaffold/stent in the presence of a maximal distal and proximal lumen diameter between 2.5 and 3.3 mm, and a 2.5 mm stent/scaffold between 2.0 and 3 mm.
As for the scaffold deployment, the pressure increment should be no more than two atm per five seconds and, once the maximal pressure is achieved, the pressure should be maintained for 30 seconds. If post-dilatation is required (recommended in case of >10% residual stenosis), a low-profile, high-pressure, non-compliant balloon should be used and inflated within the expansion limits of the selected scaffold. The size of the non-compliant balloon should not be larger than 0.25 mm more than the scaffold diameter. Multiple scaffolds or stents can be implanted into the culprit lesion. Scaffolds may be implanted abutting or overlapping. In case of the latter, the overlapping zone should not extend to more than 4 mm.
PERIPROCEDURAL MEDICATION
The use of bivalirudin is recommended for periprocedural anticoagulation (i.v. bolus of 0.75 mg/kg followed by an infusion of 1.75 mg/kg/hr not titrated to ACT and usually terminated at the end of the procedure). Alternatively, heparin may be used (bolus of heparin of 100 U/kg weight, or 60 U/kg weight if GP IIb/IIIa antagonists are used). The use of GP IIb/IIIa antagonists is at the discretion of the operator.
ANTIPLATELET THERAPY
All patients receive a loading dose of aspirin (250-300 mg i.v.) and a potent P2Y12 inhibitor (loading dose of either prasugrel 60 mg or ticagrelor 180 mg p.o.). The use of either prasugrel (10 mg daily) or ticagrelor (90 mg twice daily) is mandatory for the first month. Thereafter, a conversion to clopidogrel is allowed, with the aim of maintaining dual antiplatelet therapy throughout 12 months.
PRIMARY ENDPOINT
The primary endpoint is the neointimal healing score assessed at six months. The neointimal healing score is based on four scaffold- or stent-related characteristics as shown in Figure 2 and is calculated on a lesion level17,18.
1. Presence of filling defect (% intraluminal defect, ILD) is assigned a weight of “4”
2. Presence of both malapposed and uncovered struts (% malapposed/uncovered, MU) is assigned a weight of “3”
3. Presence of uncovered struts alone (% malapposed, M) is assigned a weight of “2”
4. Presence of malapposed struts alone (% uncovered, U) is assigned a weight of “1”
Neointimal healing score=(%ILD*4)+(%MU*3)+(%U*2)+(%M*1).
For endpoint analysis, stent/scaffold area and derived measures are based on the abluminal stent/scaffold contour. The healing score is calculated per lesion. The %ILD is calculated as a percentage of intraluminal defect volume divided by the scaffold/stent volume. %MU, %U and %M are calculated as a percentage of malapposed and/or uncovered struts divided by the total number of struts in the lesion.
In order to address the excessive neointimal formation as a sign of delayed healing, the modified healing score is used as one of the secondary endpoints. The value of the percentage volume obstruction (%VO) over 30% was added to the healing score and assigned a weight of 1.
The modified healing score is calculated as: (%ILD*4)+(%MU*3)+(%U*2)+(%M*1)+(max(0,%VO>30)*1).
RATIONALE FOR PRIMARY ENDPOINT
The neointimal healing score aims to assess morphological characteristics, which may be associated with impaired clinical outcomes including predisposition to scaffold/stent thrombosis. As intraluminal mass represents thrombus (alternatively fibrin), it received the largest weight. Malapposed and uncovered stent struts have been identified to correlate with stent thrombosis in autopsy7 and intravascular imaging studies19, and received a high weight when observed in the same strut and a lower weight when they occurred individually. Restenosis can lead to a thrombo-restenotic phenomenon and assumed the lowest weight (only added in the modified healing score, which is a secondary endpoint). No prospective validation of the healing score is available so far, with the exception of data analysed for the purpose of the power calculation18 for this study, as mentioned in the statistical paragraph.
RATIONALE FOR TIME POINT OF PRIMARY ENDPOINT ASSESSMENT
The neointimal healing score is evaluated at six months for several reasons. It is of particular interest to know the healing response, including scaffold/stent coverage and malapposition, prior to 12 months in view of unforeseen DAPT cessation in a potentially vulnerable environment. In addition, the peak of neointimal hyperplasia occurs between three and six months and therefore justifies the assessment at six months. The large thrombus burden present in STEMI lesions may impact on the healing response by thrombus dissolution, an effect that is expected to be evident during the first months after primary PCI.
SECONDARY ENDPOINTS
Secondary clinical endpoints are device-oriented MACE (cardiac death, MI not clearly attributable to a non-intervention vessel, and clinically indicated TLR) at one, six, and 36 months (Online Appendix). Additional secondary endpoints include the individual components of device-oriented MACE as well as all-cause death, any MI, non-clinically indicated TLR, clinically indicated and non-clinically indicated target vessel revascularisation, scaffold/stent thrombosis according to the Academic Research Consortium definition, and angina status. Recurrent MI is defined according to the Third Universal Definition of MI as evidence of myocardial necrosis in a clinical setting consistent with acute MI, as specified in the Online Appendix. The definition of acute device and procedure success is provided in the Online Appendix.
All clinical events are adjudicated by an independent clinical events adjudication committee.
Angiographic endpoints at six months include percent diameter stenosis (% DS), minimal lumen diameter (MLD), late lumen loss (LL) and binary restenosis. All angiographic endpoints will be assessed at the in-segment, in-device, proximal and distal region.
Secondary OFDI endpoints are assessed at six months and include all individual components of the neointimal healing score, the mean and minimal scaffold/stent and diameter, area and volume, the frequency of incomplete strut apposition including area and volume, the percentage of uncovered struts, the mean thickness of strut coverage and neointimal hyperplasia area and volume, the mean flow area and volume and intraluminal defect area and volume.
OPTICAL FREQUENCY DOMAIN IMAGING
OFDI will be performed at six-month follow-up using the Lunawave® Coronary imaging console (Terumo Corp., Tokyo, Japan) and the FastView catheter (Terumo Corp.) over a 6 Fr guiding catheter. Flushing rates amount to 3-4 ml/sec in the RCA and 4-5 ml/sec in the left coronary system. Heparin will be administered to achieve an activated clotting time >250 sec. The FastView catheter will be advanced over a coronary wire at least 5 mm distal to the study lesion. After administration of 100-200 µg intracoronary nitroglycerine, the pullback will be acquired at a speed of 40 mm/sec. In case of an early revascularisation of the study scaffold/stent within three months after implantation, only the angiographic endpoints will be assessed. In case of an intercurrent angiography occurring more than three months after scaffold/stent implantation, OFDI and angiography will be performed for the primary and secondary endpoint assessment.
OFDI recordings will be sent to an independent core laboratory (Cardialysis B.V., Rotterdam, The Netherlands) for analysis. It is not possible to blind the analysts to the stent type based on the characteristic appearance of Absorb BVS and EES stent struts. Taking into account the difference in the optical properties of cobalt-chromium and polylactide, OFDI analysis will be performed using comparative methods.
At two centres, flow measurements at rest and under hyperaemia will be performed on the occasion of the six-month follow-up (fractional flow reserve, microcirculatory resistance and coronary flow reserve) to investigate differences between devices.
DEFINITIONS OF OFDI-DEFINED NIH COMPONENTS
INTRALUMINAL MASS
An irregularly shaped structure in contact with the luminal contour is defined as an intraluminal mass attached to the vessel wall. The area of this defect can be measured. An isolated structure in the lumen without contact to the vessel wall is defined as a free intraluminal mass.
MALAPPOSITION
A strut is defined as malapposed when the distance between the endoluminal surface of the strut, with respect to the interpolated lumen contour, is more than the strut thickness of either a metallic or a polymeric strut. It is measured at the midpoint of the endoluminal reflective border of metallic stents or the endoluminal side of the reflective frame of polymeric scaffolds. The malapposition distance is the distance between the interpolated lumen contour and the reverse of the completely malapposed metallic or polymeric struts (the reverse of the bright reflective frame in polymeric struts, and virtual reverse [=91 um from the bright leading edge] in metallic [XIENCE] struts) at the midpoint of the endoluminal edge of the strut. A malapposition distance greater than zero is the criterion for malapposition in this study. At follow-up, in polymeric scaffolds, the abluminal side of the black core is used for the measurement of malapposition distance, since the abluminal reflective bright frame cannot be distinguished from the neointima coverage.
COVERAGE
In metallic DES, the struts are classified as covered in the presence of a coverage thickness >0 μm (tissue can be identified above the struts) and ≥30 μm in polymeric struts. The 30 μm threshold is derived from the thickness of the endoluminal bright border of the strut core without any coverage immediately after the procedure3.
DATA CAPTURE AND MONITORING
Data will be entered into a web-based CRF. Data entry will be monitored according to a pre-specified monitoring plan.
STUDY ORGANISATION
The trial was designed by the principal investigators. The trial was sponsored by the European Cardiovascular Research Institute and supported with an unrestricted grant from Terumo and Abbott Vascular. The independent clinical events committee is responsible for adjudicating all cardiovascular events. The independent core laboratory is responsible for all OFDI and angiography related analyses.
Data management
All data are collected by electronic case report forms. Participating centres are responsible for data entry. Data management and monitoring are performed by the contract research organisation. A 100% source data verification is performed for the first two patients enrolled at each site. Subsequent patients are monitored as outlined in the study-specific monitoring plan. Special attention is paid to the six-month (OFDI) data as this is the primary endpoint of the study. Based on the findings (e.g., protocol deviations for informed consent procedure, SA reporting, etc.) the degree of monitoring can be increased (risk-based monitoring).
Statistical analysis
POWER CALCULATION
We assume a mean neointimal healing score of 9.0 in the Absorb BVS scaffold group with a standard deviation of 9.4. These data are based on an OFDI analysis of the ABSORB Cohort B1. The healing score of the EES is anticipated to be similar to the one observed with the Absorb BVS (with similar SD) based on historical 12-month data showing a healing score of 10.8 (SD 15.3) (data on file). With a non-inferiority margin of 4.5 points, a one-sided significance level (alpha) of 0.05 and an attrition rate of 20%, 190 patients will provide 90% power to confirm non-inferiority. If the upper confidence bound does not exceed the non-inferiority margin (+4.5 points) then the Absorb BVS will be considered to be non-inferior to EES.
The primary endpoint and all imaging-based (OFDI and angiography) findings will be analysed based on the as-treated principle. Clinical outcomes will be based on the intention-to-treat principle.
Conclusions
This will be the first randomised study investigating arterial healing in response to implantation of the Absorb BVS compared with the EES in the setting of STEMI. A novel primary endpoint –the neointimal healing score– is assessed at six months using optical coherence tomography. The arterial healing response, in conjunction with results on angiographic efficacy parameters and device-oriented MACE, will elucidate disease-specific applications of bioresorbable scaffolds and provide an important basis for future clinical investigations of BVS in STEMI patients.
| Impact on daily practice This will be the first randomised study assessing the arterial healing response of the Absorb BVS versus the XIENCE metallic DES by means of a neointimal healing score among STEMI patients. The neointimal healing score may be associated with clinical outcomes including predisposition to scaffold or stent thrombosis and therefore provide relevant insights into the projected safety and efficacy of Absorb BVS in primary PCI. |
Guest Editor
This paper was guest edited by Giulio Guagliumi, MD; Division of Cardiology, Ospedali Riuniti di Bergamo, Bergamo, Italy.
Conflict of interest statement
S. Windecker has received research contracts to the institution from Abbott. The other authors have no conflicts of interest to declare. The Guest Editor is a consultant for St. Jude Medical and Boston Scientific and has received research grants through the hospital from St. Jude Medical and Boston Scientific.
Online Appendix. Clinical endpoint definitions
A. MYOCARDIAL INFARCTION
MI is defined according to the Third Universal Definition of Myocardial Infarction as evidence of myocardial necrosis in a clinical setting consistent with acute myocardial ischaemia.
1. SPONTANEOUS MI (>48 HOURS AFTER INTERVENTION)
Q-wave MI
Development of new pathological Q-waves in two or more contiguous leads (as assessed by the ECG core laboratory) with or without post-procedure troponin, CK or CK-MB levels elevated above normal.
Non-Q-wave MI
Detection of a rise and/or fall of cardiac biomarker values (preferably cardiac troponin) with at least one value above the 99th percentile upper reference limit (URL) and with at least one of the following:
– Symptoms of ischaemia.
– New or presumed new significant ST-segment-T-wave (ST–T) changes or new left bundle branch block (LBBB).
– Development of pathological Q-waves in the ECG½1
– Imaging evidence of new loss of viable myocardium or new regional wall motion abnormality.
– Identification of an intracoronary thrombus by angiography or autopsy.
2. PERIPROCEDURAL MI AFTER PCI (WITHIN 48 HOURS AFTER PCI)
Elevation of troponin values >5x99th percentile URL
in patients with normal baseline values (≤99th percentile URL).
3. PERIPROCEDURAL MI AFTER CABG (WITHIN 48 HOURS AFTER CABG)
Elevation of troponin values >10x99th percentile URL
in patients with normal baseline values (≤99th percentile URL).
In addition, at least one of the following is required:
– New pathological Q-waves or new LBBB, or
– Angiographic documented new graft, or
– Native coronary artery occlusion, or
– Imaging evidence of new loss of viable myocardium or new regional wall motion abnormality.
Note: if troponin not available, CK or CK-MB will be used (in hierarchical order: CK, CK-MB) with the same thresholds as described for troponin.
Note: important: #2 and #3 are only applicable for periprocedural MIs during re-interventions (i.e., not applicable for periprocedural MIs during the index procedure).
4. REINFARCTION - BASELINE BIOMARKERS OF MYOCARDIAL DAMAGE: CK, CK-MB, AND/OR TROPONIN >1xURL OR ACUTE MI IN PROGRESS
Scenario A) CK (or CK-MB) from index MI has not yet reached its maximum level: recurrent thoracic chest pain or ischaemia equivalent >20 minutes (or new ECG changes consistent with MI) and appropriate cardiac enzyme data: 1) a rise in CK within 24 hours of the index event >2*URL (confirmed by either CK-MB or troponin >1*URL) and ≥50% above the previous level, or 2) in the absence of CK: a (post-PCI) rise in CK-MB within 24 hours of the index event >3*URL and ≥50% above the previous level, or 3) in the absence of CK and CK-MB: a (post-PCI) rise of troponin within 24 hours of the index event >3*URL and ≥50% above the previous level.
Scenario B) Elevated CK (or CK-MB) following the index MI has peaked and CK level has returned to
Scenario C) CK (or CK-MB) when the index MI has peaked and CK level has not returned to
TARGET VESSEL VS. NON-TARGET VESSEL MI
Any MI not clearly attributable to a non-target vessel will be considered as target vessel MI.
Note: important: staged lesion(s) are non-study lesion(s). Therefore, MIs related to staged lesion(s) cannot be target vessel MIs.
B. REVASCULARISATION
The revascularisations will be adjudicated as per the ARC definition.
LOCATION OF REVASCULARISATION:
Target lesion revascularisation (TLR)
TLR is defined as any repeat percutaneous intervention of the target lesion or bypass surgery of the target vessel performed for restenosis or other complication of the target lesion. The target lesion is defined as the treated segment from 5 mm proximal to the stent and up to 5 mm distal to the stent/scaffold.
Target vessel revascularisation (TVR)
TVR is defined as any repeat percutaneous intervention or surgical bypass of any segment of the target vessel. The target vessel is defined as the entire major coronary vessel proximal and distal to the target lesion which includes upstream and downstream branches and the target lesion itself.
Non-target lesion revascularisation (Non-TLR)
Any revascularisation in the target vessel for a lesion other than the target lesion is considered a non-TLR.
Non-target vessel revascularisation (Non-TVR)
Revascularisation of the vessel identified and treated as the non-target vessel at the time of the index procedure.
Note: TLR and TVR will be adjudicated by the angiographic core laboratory.
Note: important: staged lesion(s) are non-study lesion(s). Therefore, revascularisations related to staged lesion(s) cannot be target lesion revascularisations.
Clinically indicated revascularisation (CI-TLR/TVR)
A revascularisation is considered clinically indicated if associated with any of the following:
– Positive functional ischaemia study including positive FFR
– Ischaemic symptoms and angiographic diameter stenosis ≥50% by core laboratory QCA
– Angiographic diameter stenosis ≥70% by core laboratory QCA without angina or positive functional study
CORONARY ARTERY BYPASS GRAFT SURGERY
CABG during follow-up is only considered as a clinically indicated target lesion revascularisation if coronary angiography indicates a diameter of stenosis ≥50% of the index lesion (core lab QCA assessment) associated with one of the following conditions:
– A positive history of recurrent angina pectoris presumably related to the target vessel
– Objective signs of ischaemia (12-lead ECG, exercise test or equivalent) presumably related to the target vessel
– Abnormal results of any invasive functional diagnostic test (e.g., Doppler flow velocity reserve, fractional flow reserve)
– A TLR/TVR with a diameter stenosis ≥70% (core lab QCA assessment) in the absence of the above-mentioned ischaemic signs or symptoms
C. ACUTE SUCCESS DEFINITION
CLINICAL DEVICE SUCCESS (LESION BASIS)
Successful delivery and deployment of the assigned device at the intended target lesion and successful withdrawal of the delivery system with attainment of final in-scaffold/stent residual stenosis of <30% by QCA (by visual estimation if QCA unavailable).
CLINICAL PROCEDURE SUCCESS (PATIENT BASIS)
Achievement of final in-scaffold/stent residual stenosis of <30% by QCA (by visual estimation if QCA unavailable) with successful delivery and deployment of the assigned device at the intended target lesion and successful withdrawal of the delivery system without the occurrence of DoCE during the hospital stay (maximum of seven days), and with or without use of other therapeutic devices.
