Abstract
Aims: Genes encoding vascular endothelial growth factor (VEGF) can potentially augment myocardial perfusion in patients with coronary artery disease (CAD). We conducted a randomised, double-blind, placebo-controlled gene therapy study with the adenovirus carrying VEGF121 (BIOBYPASS [AdGVVEGF121.10NH]).
Methods and results: Seventeen patients with severe CAD were 2:1 randomised to BIOBYPASS (n=12; 61 years) or placebo (n=5; 64) as 12 intra-myocardial injections into the ischaemic area using the NOGA XP® system. The study was terminated prematurely due to a company product portfolio decision. Mean change in total exercise duration from baseline to 12, 26 and 52 weeks was 20.2, 21.4 and 16.4 sec in BIOBYPASS treated and 46.2, 31.4 and 12.4 sec in placebo (NS). Change from baseline to at least 1 mm ST depression during exercise at 12, 26 and 52 weeks did not differ between BIOBYPASS and placebo. Change in stress-induced ischaemia score was similar in the BIOBYPASS (3.4%) and placebo (2.0%) groups. An improvement in symptoms was seen in patients treated with BIOBYPASS, but no difference between the groups.
Conclusions: Direct intramyocardial injection of BIOBYPASS (AdGVVEGF121.10NH) was safe but did not improve exercise capacity, time to ischaemic threshold or myocardial perfusion compared to sham injection in patients with refractory myocardial ischaemia.
Introduction
Within the last two centuries, the development of and progress in modern cardiovascular drug therapies and mechanical revascularisation with balloon angioplasty and coronary artery bypass surgery has improved the prognosis for patients with both acute and chronic myocardial ischaemia. However, there are many patients with severe coronary artery disease (CAD), who cannot be treated satisfactorily with conventional therapies. This has lead to an extensive research to find new treatment modalities. The intensive research within the field of molecular biology has discovered several families of proteins with an angiogenic potential1,2. The vascular endothelial growth factors and the fibroblast growth factors have both the potential to induce therapeutic angiogenesis, i.e., growth of new vessels, in the human myocardium and they have both been tested in patients with CAD1,2.
In several small clinical studies, intramyocardial or intracoronary gene transfer of VEGF or FGF demonstrated clinical evidence of improved myocardial perfusion in patients with severe CAD3-7. In the first large double-blind placebo-controlled study (EUROINJECT ONE) with plasmid VEGF-A165 gene transfer, improvement in local wall motion in the treatment area in the active treated group was detected compared to the placebo group8,9. The myocardial perfusion improved in the VEGF group but not in the placebo group. However, there was no clinical effect as demonstrated in earlier smaller safety trials3-7. Moreover, in a recent study with a four times higher plasmid VEGF-A165 gene dose as used in the EUROINJECT ONE trial, it was not possible to detect any improvement in myocardial perfusion or clinical symptoms10. The use of an adenoviral vector for transfection may increase the gene transfection rate. Thus, this treatment modality using adenovirus-5-VEGF-121 (AdV5-121) was tested in a small safety and feasibility study in patients with stable refractory angina2. Based on these results, the larger REVASC trial with AdV5-121 was initiated and finalised11,12. These two unblinded studies demonstrated the safety of direct intramyocardial injections of AdV5-121 and efficacy in terms of improved exercise time and reduced symptoms compared to a medically treated control group. These encouraging results were the basis for the design of the present double-blind, placebo- controlled study of the safety and efficacy of BIOBYPASS (AdGVVEGF121.10NH) gene therapy in patients with refractory advanced CAD – the NOVA trial. The study was initiated April 2005 and terminated prematurely February 2007 due to the slow inclusion rate, which led to a company product portfolio decision by GenVec Inc. (Gaithersburg, MD, USA). The present paper describes the safety and efficacy data for the 17 patients included.
Materials and methods
The NOVA study was designed as a randomised, double-blind, placebo-controlled, Phase II multicentre trial in Europe and Israel initiated by GenVec Inc, USA and Cordis Corporation (Johnson & Johnson, Warren, NJ, USA). The trial was approved by the regional Ethics Committee and Medicines Agencies in the participating countries and was conducted according to the Declaration II of Helsinki. The patients were included after oral and written information about the study, and after signing a written informed consent. The study has been registered in clinicaltrials.gov (NCT00215696).
The study was planned to include 129 patients with advanced CAD and moderate to severe angina pectoris despite optimal medical treatment, who were not amenable to percutaneous coronary revascularisation or coronary bypass grafting in the judgment of an independent cardiac surgeon. Inclusion criteria were: more than 10% reversible ischaemia of left ventricle at an adenosine stress single photon emission computerised tomography (SPECT), a coronary arteriography demonstrating at least one main coronary vessel from which new collaterals/vessels could be supplied, age between 18 and 80 years, Canadian Cardiovascular Society angina classification (CCS) ≥2 in spite of optimal anti-angina medicinal therapy, two baseline bicycle exercise tolerance tests (ETTs) performed meeting the following criteria (exercise time between two and eight minutes until angina level 3 and exercise duration on the two ETTs should be within 15% of each other).
Excluded were patients with unstable angina pectoris, acute myocardial infarction within the last six weeks, left ventricular ejection fraction below 25%, NYHA 3–4, diabetes mellitus with proliferative retinopathy, diagnosed or suspected cancer disease, chronic inflammatory disease, fertile women, and patients who had received VEGF or any other angiogenic agent or gene therapy in the past. According to the decisions of cardiac surgeons and cardiologists none of the patients could be treated further by conventional revascularisation.
Study protocol
After screening, the patients were randomised in a 2:1 fashion to receive BIOBYPASS (4x1010 pu) or placebo administered by direct intramyocardial injection using the NOGA-guided Myostar® (Biologics Delivery Systems, Johnson & Johnson, Warren, NJ, USA) percutaneous injection catheter system. The patients were then followed for 12 months to evaluate the safety and efficacy of the treatment. The patients were followed once a week the first months and then at week 12, 26 and 52 to control for drug related adverse events and safety issues. Prescriptions of anti-angina or vasoactive medications were not changed during the study period.
To evaluate the effect of the treatment the patients underwent in the follow-up period bicycle exercise tolerance testing (ETT), registration of angina pectoris class according to the CCS classification after 12, 26 and 52 weeks and SPECT after 26 weeks.
Endpoints
The primary endpoint was the change from baseline in total exercise duration on ETT at week 26.
Secondary endpoints were change of : I) reversible perfusion defect size in SPECT perfusion study at week 26; II) total exercise duration at weeks 12 and 52 and III) time-to-onset of 1 mm ST depression on ETT, and CCS class, frequency of angina attacks and nitroglycerine (NTG) consumption per two weeks at week 12, 26 and 52.
BIOBYPASS
BIOBYPASS (AdGVVEGF121.10NH) is a modified version of the adenovirus serotype 5 (Ad5) genome deleted of E1a, E1b and the majority of E3 sequences, rendering it replication deficient. The deleted E1a sequences have been replaced with the VEGF121 transgene downstream from a CMV promoter. The diluent (FFB) used to suspend AdVEGF consists of polysorbate 80 (0.025 g/L), sodium chloride (4.383 g/L), magnesium chloride (0.203 g/L), Tris HCl, pH 7.6±0.2 (1.211 g/L), trehalose (55.25 g/L), and WFI (q.s. to 1.0 L). Study treatment was supplied in double-blind packaging, in 4 mL vials containing 1.7e10 pu/mL AdVEGF in FFB, or FFB only.
NOGA® XP – electromechanical mapping of left ventricle
Electromechanical evaluation of the left ventricle was performed with the NOGA® XP system (Biologics Delivery System, Johnson & Johnson) as previously described6,7,13,14. The diagnostic NOGA® catheter was introduced percutaneously via the groin into the left ventricle. A sensor at the tip of the catheter, in contact with the endocardial surface, registered the local myocardial unipolar voltage (UVI) and regional wall motion/contraction (local shortening [LS]) within the left ventricle. NOGA creates a three-dimensional colour image of the left ventricle.
Intramyocardial injections
The intramyocardial injections were performed with the 8 Fr-sized Myostar® mapping-injection catheter (Biologics Delivery System, Johnson & Johnson) inserted into the left ventricle via the groin6,7,13-15. Twelve 0.2 ml injections were given around and within the area with reversible ischaemia with a total dose of BIOBYPASS (4x1010 pu) or placebo. The injections were performed slowly (30-40 sec) and only to areas with unipolar voltage above 5 mV and with a thickness of the ventricle wall on echocardiography exceeding 6 mm. Quality control of the injections included: 1) the catheter’s tip perpendicular (±300) to the left ventricular wall in two planes, 2) the loop stability at the same level as during the mapping procedure, if possible <2 mm, and 3) one or more ectopic extra-ventricular beats should appear in the exact moment of the protrusion of the injection needle into the myocardium.
Single photon emission computerised tomography (SPECT)
SPECT studies were performed as a 2-day protocol (500-700 MBq 99mTc-sestamibi at each study) with adenosine infusion over
4-6 minutes (0.14 mg/kg/min by infusion pump), if possible combined with a sub-maximal exercise test as previously described6. Care was taken to perform the stress test at the inclusion and at the follow-up studies with identical cumulative adenosine doses and identical sub-maximal exercise loads. Gated (eight frames) imaging was performed with a two-headed Millennium GE gamma camera (GE Healthcare Ltd, Little Chalfont, Buckinghamshire, UK), with a gadolinium interleaved attenuation-scatter correction.
Power calculation
The power calculation was based on the experience from the REVASC study, where the variability of delta ETT was found to be 1.6 min. Based on this result the ETT variability to be used for the sample size calculation in the NOVA study was decided to be 1.8 min. With a 2:1 randomisation, 80 % power, alpha=0.05, ETT SD of 1.8 min, and an estimated difference in ETT time between BIOBYPASS-treated and placebo group of one minute, the calculated needed number of participants treated was 120. Another nine patients would be enrolled to compensate for the expected 7% of patients to be lost to follow-up. Thus, the total number of patients in the study would be 129.
Statistics
Data are presented as mean±standard error of the mean. Continuous variables were compared using Student’s t test and categorical variables were compared using Fisher’s exact test. The one-way ANOVA was used for multiple variables comparison. The comparisons between baseline and follow-up data measured within patient’s group by the Wilcoxon signed-ranks test for paired samples. The dependent/outcome variable in the ANCOVA model, which was the change from baseline in total exercise duration or time to onset where the baseline value was included as a covariate to increase the precision in determining the treatment effect (BIOBYPASS versus placebo). The ANCOVA model also included a term for the effect of treatment (i.e., BIOBYPASS) and was conducted using the intension to treat population.
All data were analysed using SPSS statistical analysis program (SPSS version 11.0; SPSS INC., Chicago, IL, USA). Values of P<0.05 were considered significant.
Results
A total of 17 patients were included and treated in four centres before termination of the study, where 12 patients had been treated with BIOBYPASS and five with placebo. The demographic and clinical characteristics of the included patients treated with BIOBYPASS or placebo are outlined in Table 1.
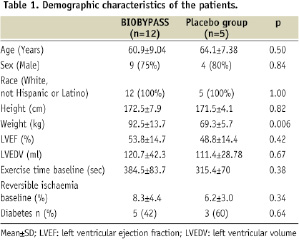
All patients had severe angina pectoris, limited exercise capacity and reversible ischaemia on SPECT. The weight was significantly higher in patients treated with BIOBYPASS compared to placebo patients.
BIOBYPASS treatment did not increase total exercise time or time to 1 mm ST-depression 12, 26 and 52 weeks after treatment compared to placebo (Table 2 and Figure 1).
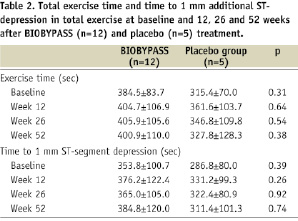
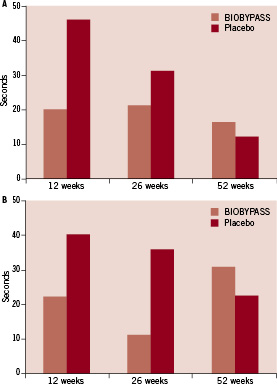
Figure 1. Changes in total exercise time (A) and time to 1 mm additional ST-depression (B) from baseline to 12, 26 and 52 weeks after BIOBYPASS (n=12) and placebo (n=5) treatment
The degree of reversible ischaemia on SPECT was identical in the two groups at baseline and there was no improvement in absolute or change in perfusion at 26 weeks follow-up in any of the groups (Table 3 and Figure 2).
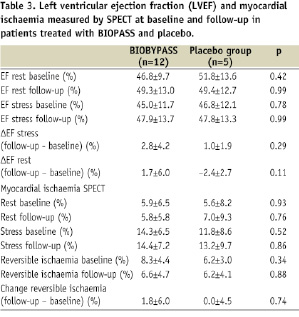
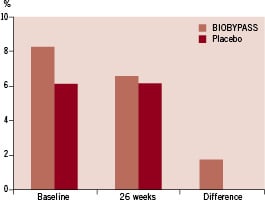
Figure 2. The total amount of reversible ischaemia of the left ventricle at baseline and follow-up, and the calculated difference from baseline to 26 weeks after BIOBYPASS (n=12) and placebo (n=5) treatment
The ventricular function at baseline was not different between BIOBYPASS and placebo and no improvement was detected in rest and stress ejection fraction at follow-up (Table 3).
BIOBYPASS treated patients had significantly reduced angina CCS classification, angina frequency and nitroglycerine consumption 12, 26 and 52 weeks after treatment (Table 4).
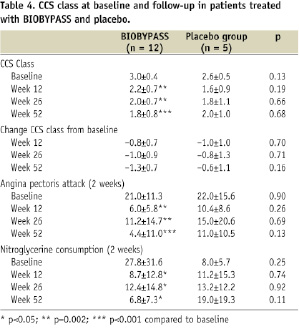
The same trend was not seen in placebo-treated patients. However, there was no difference between change in CCS classification between BIOBYPASS and placebo-treated patients (Table 4 and Figure 3).
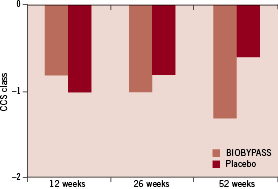
Figure 3. The changes in CCS class from baseline to 12, 26 and 52 weeks after BIOBYPASS (n=12) and placebo (n=5) treatment.
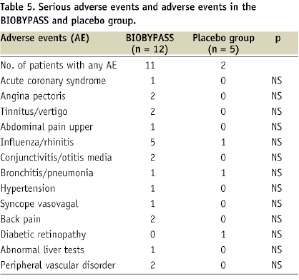
BIOBYPASS was considered potentially associated with a single (8.3%) serious drug-related adverse event of transient moderate fever. The overall number of patients with any adverse event considered to be drug-related within one year follow-up was four (33%) in the BIOBYPASS group vs. one (20%) in the placebo group; including pyrexia (16.7%), influenza-like symptoms (8.3%), liver function test abnormality (8.3%) and subcutaneous erythema (8.3%). All registered adverse events are outlined in Table 5 and they were all reversible.
Discussion
There are only a few clinical studies published regarding gene therapy of patients with coronary artery disease. This is a presentation of the results of a randomised, double-blind, placebo-controlled study to test the safety and efficacy of gene therapy using an adenovirus vector encoding VEGF 121 (BIOBYPASS). Although the study was terminated before the final number of patients had been included due to a company product portfolio decision, we feel that it is important to publish the safety and efficacy data for the included 17 patients for the planning of future clinical gene therapy studies. These study results should be interpreted cautiously due to the small number of patients included.
There was no improvement in total exercise capacity, time to 1-mm ST-depression, degree of reversible ischaemia on SPECT or left ventricular ejection fraction at follow-up in BIOBYPASS treated compared to placebo patients. These results are in contrast to the previously published data from the open REVASC study with BIOBYPASS, where there was a significant improvement in total exercise capacity and a trend toward reduced reversible ischaemia on SPECT12. In the EUROINJECT ONE with 500 μg plasmid VEGF-A165 gene treatment, it was possible to detect an improvement in myocardial perfusion in the VEGF group but not in the placebo group8,9. In addition, there was an improvement in local wall motion in the treatment area in the active treated group compared to the placebo group. Interestingly, this was not transferred into a clinical effect in the patients. The explanation could be a too low transfection rate when using a low plasmid VEGF-A165 dose. However, in a recent published study using a four times higher plasmid VEGF-A165 dose (2,000 μg) Stewart et al, 200910 could not detect any effect of the treatment on any of the endpoints in the study. This indicates that a more effective transfection rate using i.e., adenovirus or transfected cells could be needed to induce neovascularisation in ischaemic myocardium.
The patients treated with BIOBYPASS had significantly reduced angina, frequency of angina attacks and nitroglycerine (NTG) consumption 12, 26 and 52 weeks after treatment compared to baseline. No improvements were detected in placebo treated patients. However, when comparing the BIOBYPASS and placebo treated patients, there was no difference in angina between the two groups. This could be due to the small number of patients included in the study, since the REVASC study demonstrated reduced angina in the active treated patients compared to the control group.
The study was without any complications caused by the delivery method with direct intramyocardial injection of the BIOBYPASS or placebo. This is in agreement with a recent study focusing on the safety of direct intramyocardial injections using the NOGA XP technology13.
There were only few of the expected short-time reversible side-effects to the treatment with an adenovirus such as, fever, influenza-like symptoms and liver affection, which not was different from the safety data in the REVASC study12. There was no other sign clinically or biochemically of a systemic effect of the adenovirus treatment. The treatment was performed without any special in-hospital safety concerns and the used materials handled as routine infected materials from patients.
The treatment was therefore considered to be safe and feasible for clinical use. In the post hoc combined analysis of the two studies AGENT 3 and 4 with intravenous injections of Ad5FGF-4 in patients with stable chronic myocardial ischaemia there was no effect of the treatment on exercise or clinical parameters. However, when dividing the patients into genders, there was a clear difference in response between men and women. Men did not respond to the treatment, but there was a clear effect of treatment with Ad5FGF-4 in women. They had increased total exercise time, time to 1 mm ST-depression, time to angina and a reduction in CCS class16. It is very interesting since it is the first time that a gender specific difference in response has been discovered in cardiac angiogenic therapy. Whether the same tendency could be present in the NOVA study is impossible to evaluate due to the small number of patients included.
The results of the present study are in line with previous unblinded or blinded placebo-controlled studies with growth factors or genes encoding growth factors in patients with chronic coronary artery disease8,9,12,14. There is presently no clear tendency in treatment strategies with different growth factors. However, it cannot be excluded that the results would have been different if the present study had been continued as planned. Moreover, the presence of adenovirus antibodies pre-treatment could potentially reduce the gene transfection efficacy. However, these antibodies were not measured in the present study.
For future studies, it has to be considered whether it is possible to improve the delivery method and the gene transfection rate to improve clinical outcome. Moreover, it has to be evaluated whether the pre-specified endpoints actually can be measured by the methods used: i.e., is the SPECT method sensitive enough to detect the relatively small changes in perfusion the treatment is expected to induce? It is presently uncertain which of the growth factors used clinically is most effective in inducing a clinically relevant angiogenesis1,2. It is very interesting, that at recent double-blind placebo-controlled study with NV1FGF plasmid treatment in patients with critical limb ischaemia improved the clinical outcome by reducing the risk of all and major amputations17.
In conclusion, the study demonstrated that it was safe to perform direct intramyocardial injections of the BIOBYPASS or placebo in patients with stable severe coronary artery disease. However, it was not possible to detect any clinical beneficial effect of the treatment on exercise capacity, myocardial perfusion or clinical symptoms when comparing BIOBYPASS and placebo treated. This could be due to the small number of patients included.