Abstract
Aims: Titanium-nitride-oxide-coated bioactive stents (BAS) have demonstrated a favourable outcome when compared with paclitaxel-eluting stents in patients with acute myocardial infarction (MI). In a prospective randomised non-inferiority study design, we compared the safety and efficacy of BAS versus everolimus-eluting stents (EES) in patients with acute coronary syndrome (ACS).
Methods and results: We randomised 827 patients with ACS (1:1) to either BAS (417) or EES (410). The primary endpoint was a composite of cardiac death, non-fatal MI or ischaemia-driven target lesion revascularisation (TLR) at 12-month follow-up. Analyses were performed by intention to treat. At 12-month follow-up, the primary composite endpoint occurred in 9.6% of patients in the BAS group and 9.0% of those in the EES group (HR [hazard ratio] 1.04, 95% CI [confidence interval] 0.81-1.32, p=0.81, p for non-inferiority = 0.001). Non-fatal MI was significantly less frequent in the BAS as compared with the EES group (2.2% vs. 5.9%, p=0.007). However, the individual rates of cardiac death and ischaemia-driven TLR were similar between the two groups (1.9% vs. 1.0%, p=0.39, and 6.5% vs. 4.9%, p=0.37, respectively).
Conclusions: In patients presenting with ACS, BAS achieved a clinical outcome that was non-inferior to EES at 12-month follow-up.
In-stent restenosis has always been the “stumbling block” of coronary stenting, resulting in repeat revascularisation1-3. Over the last decade, the appearance of first-generation drug-eluting stents (DES) revolutionised the practice of coronary intervention, resulting in a reduction of restenosis rates by one-half to two-thirds at five-year follow-up4,5.
However, accumulating evidence from meta-analyses and registries has questioned the long-term safety of first-generation DES, raising concerns about a higher risk of late –and very late– stent thrombosis (ST), a potentially life-threatening complication6-8. Improved safety outcomes came with the second-generation DES. Everolimus-eluting stents (EES) significantly reduced late lumen loss when compared with paclitaxel-eluting stents, with lower rates of a composite outcome of safety and efficacy9.
The safety of titanium-nitride-oxide-coated bioactive stents (BAS) has been established in several reports from real-life unselected populations10,11. Titanium has a better biocompatibility as compared to stainless steel, gold or other surface coating materials, since it offers minimal toxic ion release, a fact that would reduce tissue reaction and inflammation12. The blood compatibility of titanium oxide concerning platelet adhesion and fibrinogen adsorption can be improved by the addition of nitrogen. Platelet adhesion and fibrinogen adsorption are lower for titanium-nitride-oxide than for titanium oxide13. Prospective studies comparing BAS with paclitaxel-eluting stents demonstrated improved clinical outcomes for the BAS group, both in patients with complex coronary lesions14 and in those presenting with acute myocardial infarction (MI)15.
These data encouraged us to perform a randomised non-inferiority trial comparing BAS with a second-generation DES (EES) in acute coronary syndromes (ACS).
Methods
STUDY DESIGN AND PATIENT POPULATION
The BASE-ACS trial is a prospective randomised multicentre active-treatment-controlled clinical trial, with the primary goal to evaluate non-inferiority in clinical outcome of BAS as compared with EES in patients presenting with ACS. We considered patients eligible for enrolment if they were above 18 years, presented with ACS, with at least one significant de novo lesion (defined as at least 50% diameter stenosis by visual estimation) in a native coronary artery or coronary bypass graft. ACS included unstable angina, non-ST-elevation and ST-elevation MI. All patients had persistent ischaemic-type chest pain or other acute symptoms consistent with myocardial ischaemia, at rest or with minimal exercise, lasting for more than 10 minutes. ST-elevation MI was defined by persistent ST segment elevation (at least 2 mm in two contiguous precordial leads, or at least 1 mm in two limb leads), new left bundle branch block, or new Q-waves in two contiguous leads with a rise of biochemical markers of myocardial necrosis (CK-MB and/or troponin) at least twice the normal upper limit. Non-ST-elevation ACS was defined by the presence of new/dynamic ECG changes compatible with ischaemia such as ST segment depression of at least 1 mm, transient ST segment elevation or ST segment elevation less than 1 mm, or T-wave inversion more than 2 mm, in at least two contiguous leads. Non-ST-elevation MI was distinguished by a rise of biochemical markers at least twice the normal upper limit.
We excluded patients with an unprotected left main disease or aorto-ostial lesions; allergy to aspirin, clopidogrel, heparin, glycoprotein IIb/IIIa inhibitors, or bivalirudin; active bleeding, or a significant increase in bleeding risk; stent length more than 28 mm, or stent diameter more than 4.0 mm needed; those who received thrombolytic therapy; those with planned surgery within 12 months of the index procedure unless dual antiplatelet therapy could be maintained throughout the perioperative period; and those with life expectancy of less than 12 months. Enrolled patients were randomly assigned in a 1:1 fashion to receive either BAS or EES. In case more than one stent was needed, the protocol mandated using the same type of stent in all of them.
Randomisation was generated by computer-based software, and implemented by a closed-envelope system stratified by centre in which every serial number of enrolment had a corresponding closed envelope containing the random allocation, which was opened only once the patient was eligible for enrolment. Study investigators were by necessity not blinded to treatment allocation; however, those who performed data management and analysis, and patients, were blinded.
The trial was initiated by the investigators, and designed by the principal investigators (PK, AY, and OH) who had unrestricted access to the data after the database was locked, made the decision to submit the manuscript for publication, revised all drafts of the manuscript, and vouch for the integrity of the trial, and the completeness and accuracy of the reported data. Participating centres were selected on the basis of their capability to execute the procedures and undertake the necessary follow-up. Before enrolment, an informed written consent was obtained from each patient after full explanation of the study protocol. The study protocol was reviewed and approved by the Ethics Committees of all the participating centres as it conforms to the ethical guidelines of the 1964 Declaration of Helsinki, as revised in 2002. The BASE-ACS trial was registered with ClinicalTrials.gov, number NCT00819923.
DEVICE DESCRIPTION
BAS (Titan2®; Hexacath, Paris, France) is a thin-strut (91 µm) balloon-expandable stent, made of stainless steel, and coated with titanium-nitride-oxide. The coating process is performed by plasma-enhanced vapour deposition of titanium in a pre-specified gas mixture of nitrogen and oxygen in a vacuum chamber. Titanium-nitride-oxide is coated on all the surfaces of the stent, both inside and outside, through a patented process which results in nitride-oxide particles on the stent surface. The stent was used in the study in diameters ranging from 2.5 to 4.0 mm, and in lengths ranging from 7 to 28 mm. EES (Xience® V; Abbott Vascular, Santa Clara, CA, USA) is a DES that contains everolimus as antiproliferative agent, at a dose of 100 µg/cm2 of stent surface area. It is coated with polyvinylidene fluoride-co-hexafluoropropylene, a fluoropolymer that elicits a biological response known as “fluoropassivation” which consists of minimising the fibrin deposition and thrombogenicity, reducing the inflammatory reaction, and enhancing a faster neointimal healing16. The total thickness of the strut and polymer is 95 µm. EES is designed to release 80% of the everolimus in the first 30 days after deployment. It was used in the study in diameters ranging from 2.5 to 4.0 mm, and in lengths ranging from 8 to 28 mm.
INDEX PROCEDURE
Patients already maintained on aspirin received no additional aspirin loading. Those not maintained on aspirin were pre-treated with aspirin at a loading dose of 250 mg orally or 250-500 mg intravenously during the procedure, and continued thereafter at a daily dose of at least 75-150 mg indefinitely. Oral clopidogrel was initiated at a loading dose of at least 300 mg before or immediately after the procedure, and continued thereafter at a daily dose of 75 mg. According to the protocol, patients in either group were prescribed oral clopidogrel for a minimum of six months, and thereafter for extended periods (maximum 12 months) according to the operator’s discretion.
During the procedure, low-molecular weight heparin (enoxaparin sodium) or unfractionated heparin was administered intravenously in the standard dosage recommended by the guidelines. Use of periprocedural glycoprotein IIb/IIIa inhibitors or bivalirudin was left up to the operator’s discretion.
In non-ST-elevation ACS, it was recommended to perform the procedure within 72 hours of admission. In ST-elevation MI, primary (or facilitated) percutaneous coronary intervention within 24 hours of admission was recommended.
Lesions were treated according to the contemporary interventional techniques. Predilatation was left to the operator’s discretion. The operator decided the appropriate diameter of the stent to be implanted aiming at a stent:vessel ratio of 1.1:1 prior to stent placement. Stents were expanded by adjusting the balloon inflation pressure to achieve an angiographic appearance of the expanded stent slightly larger than the reference vessel segment. After stent deployment, post-dilatation was allowed as necessary at the operator’s discretion. An additional stent could be deployed in overlap with the first one in case of edge dissection, incomplete lesion coverage, or otherwise suboptimal result, always dictated by the operator’s discretion.
DEFINITIONS
Patients were prospectively followed up by means of clinic visits or telephone calls by attendant cardiologists at one, six, and 12 months following the index procedure. All patient data available from hospital records, institutional electronic database, or referring physicians, were collected in trial-specific Case Report Forms in each of the participating centres and subsequently entered into a common electronic database, which was reviewed at the end of the follow-up period.
Angiographic success was defined as successful implantation of the stent into the target lesion with residual stenosis <20% and TIMI 3 flow at the end of the procedure, in the absence of dissection or thrombosis. Stent failure was defined as the inability to deliver and deploy the study stent in the target lesion. The primary composite endpoint was major adverse cardiac events (MACE) at 12-month follow-up. MACE were defined as the first occurrence of any of the following during follow-up: cardiac death, non-fatal MI, or ischaemia-driven target lesion revascularisation (TLR). Cardiac death was defined as death from cardiovascular causes or any death without another known cause. MI was diagnosed by persistent ischaemic-type chest pain with a rise of biochemical markers of myocardial necrosis (CK-MB and troponin) at least twice the upper limit of normal lab reference. In-hospital re-MI was diagnosed by a new rise of biochemical markers (CK-MB and troponin) at least 50% above the lowest level measured previously. TLR was defined as any repeat intervention (surgical or percutaneous) to treat a significant luminal stenosis (defined as >50% diameter stenosis by visual estimation) within the stent or in the 5 mm distal or proximal segments adjacent to the stent. Revascularisation was regarded as “ischaemia-driven” if it was motivated by chest pain symptoms and/or proven myocardial ischaemia in the target vessel territory by non-invasive testing. Secondary endpoints included all-cause death, a composite of cardiac death or non-fatal MI, and definite ST at 12-month follow-up. ST was adjudicated according to the criteria of definite ST described by the Academic Research Consortium (ARC)17.
Study monitors verified all data from the Case Report Forms. MACE was adjudicated by a committee whose members were unaware of the treatment allocation when adjudication of MACE was performed, and all of them came from the same institutions which participated in the trial. A Data and Safety Monitoring Committee reviewed safety data periodically and recommended each time that the study should continue without modification.
STATISTICAL ANALYSIS
The trial was powered for testing of non-inferiority for the primary composite endpoint. Previously, there were no studies addressing the use of EES in the setting of acute MI. On the other hand, BAS demonstrated a similar rate of MACE at 12 months (~ 10%) when compared with a paclitaxel-eluting TAXUS Liberté™ stent in the TITAX-AMI trial15. Based on this finding, we estimated that the rate of primary endpoint could be 9.0% to 9.5% in the present study. Assuming a 12-month rate of the primary composite endpoint of 9.2% for both stents (non-inferiority margin of 0.050 [5.0%]), we calculated that enrolment of at least 400 patients per group would yield at least 90% power to detect non-inferiority for the primary composite endpoint (one-sided α significance level 0.05). Non-inferiority will be achieved if the upper limit of the one-sided 95% confidence interval of the difference between the study groups is less than the margin. All data were presented on the basis of the intention-to-treat principle, which included all patients who underwent randomisation, regardless of the treatment actually received. In addition, per protocol analysis for the primary endpoint was included as a sensitivity analysis.
Continuous variables were presented as mean ± SD, while categorical variables were described with absolute and relative (percentage) frequencies. Comparisons between the two groups were performed using the unpaired t-test for continuous, and the Pearson’s chi-square test or Fisher’s exact test for categorical variables. All tests were two-sided and a probability value of p<0.05 was considered statistically significant. Hazard ratios (HR) and 95% confidence intervals (CI) were derived from univariate COX models for the comparisons between the groups. Time-to-event curves were constructed on the basis of all available follow-up data with the use of Kaplan-Meier estimates, and were compared with the log-rank test. Additionally, landmark time-to-event analyses for MI were also performed. In order to identify the independent predictors of MACE, first of all univariate logistic regression was performed for each of the baseline clinical, angiographic and procedural characteristics. At a second stage, the variables significantly associated (two-sided p<0.05) with dependent variables in univariate analyses were included in multivariable analysis. Results of logistic regression were presented as odds ratio (OR) with 95% CI. All data were analysed with the use of SPSS version 1618, and SAS system for Windows version 9.1 (SAS Institute Inc., Cary, NC, USA).
Results
BASELINE CLINICAL, ANGIOGRAPHIC AND PROCEDURAL DATA
Between January 2009 and September 2010, a total of 827 patients were enrolled in 14 centres, in five European countries, and one Southeast Asian country. Patients were randomly assigned to receive either BAS (417 patients, 480 lesions) or EES (410 patients, 484 lesions). The baseline clinical characteristics were evenly distributed between the two study groups (Table 1). The mean age of the study population was 63 years; 76% were males. Complex lesions (type-B and C) had a similar frequency between the two groups (89.4% and 87.3% for BAS and EES groups, respectively, p=0.39). Angiographic success was achieved in 99.8% in both groups. Other angiographic and procedural data were similar in the two groups (Table 2). Figure 1 portrays the trial flow diagram.
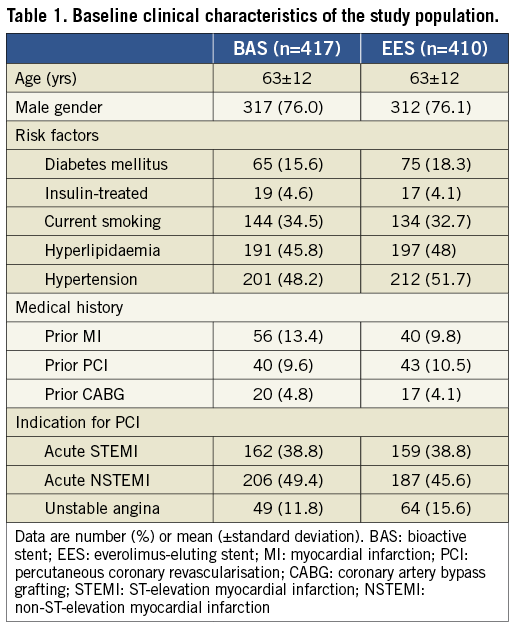
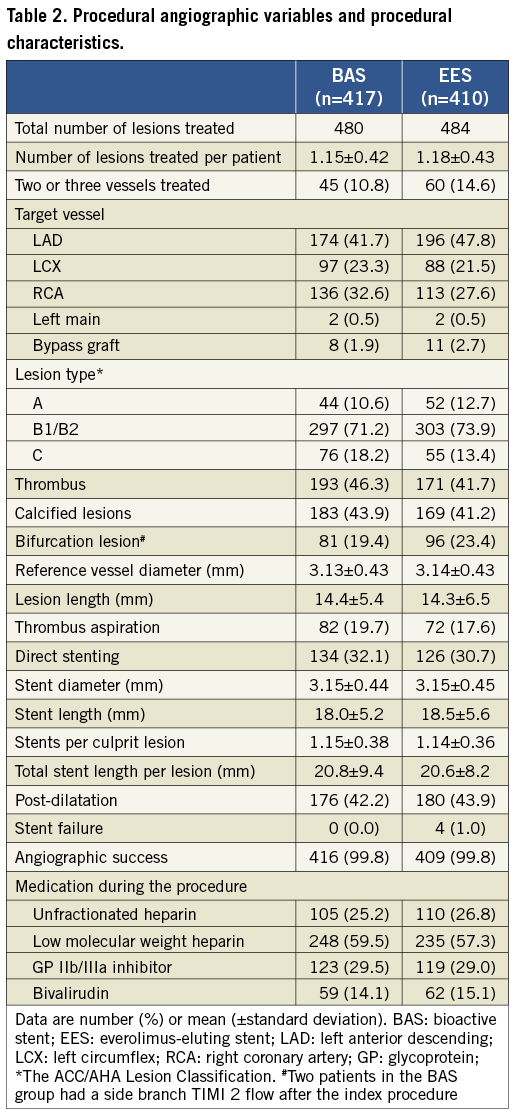
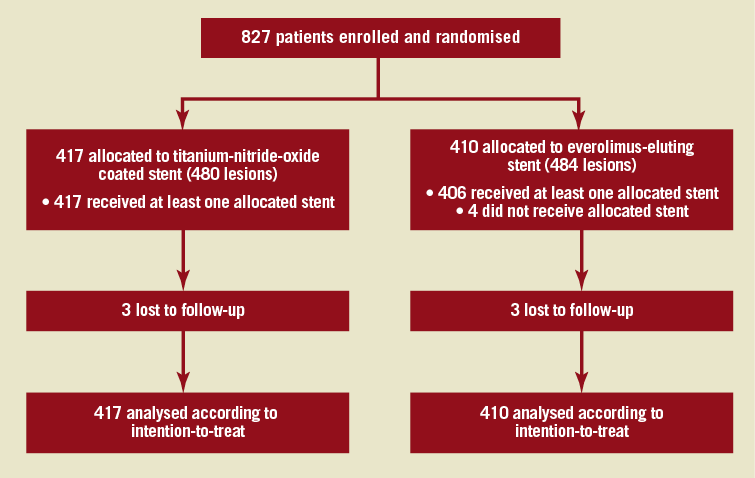
Figure 1. BASE-ACS Trial flow diagram.
CLINICAL OUTCOME
Clinical follow-up for 12 months was completed in 99.4% of patients. At six-month follow-up, 89.7% of patients in the BAS group were on clopidogrel therapy as compared with 99.3% of patients in the EES group (p<0.001). However, at 12-month follow-up, only 51.3% of patients in the BAS group were still maintained on clopidogrel as compared with 68.3% of patients in the EES group (p<0.001). The mean duration of clopidogrel use was 8.7±3.6 vs. 10.2±3.0 months, in the BAS and EES groups, respectively, (p<0.001). Clinical follow-up data are presented in Table 3.
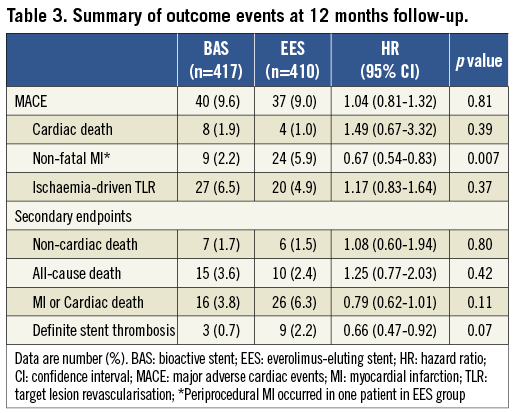
Based on the intention-to-treat principle, BAS was non-inferior, when compared with EES (Figure 2), with respect to the occurrence of the primary composite endpoint of MACE at 12-month follow-up (9.6% vs. 9.0%, respectively; HR for BAS 1.04, 95% CI 0.81-1.32, p=0.81 for superiority; p=0.001 for non-inferiority). Based on the per-protocol analysis, the primary composite endpoint of MACE at 12-month follow-up occurred in 40/417 (9.6%) patients in the BAS group, as compared with 37/406 patients (9.1%) in the EES group (HR for BAS 1.03, 95% CI 0.81-1.31, p=0.81; p=0.001 for non-inferiority).
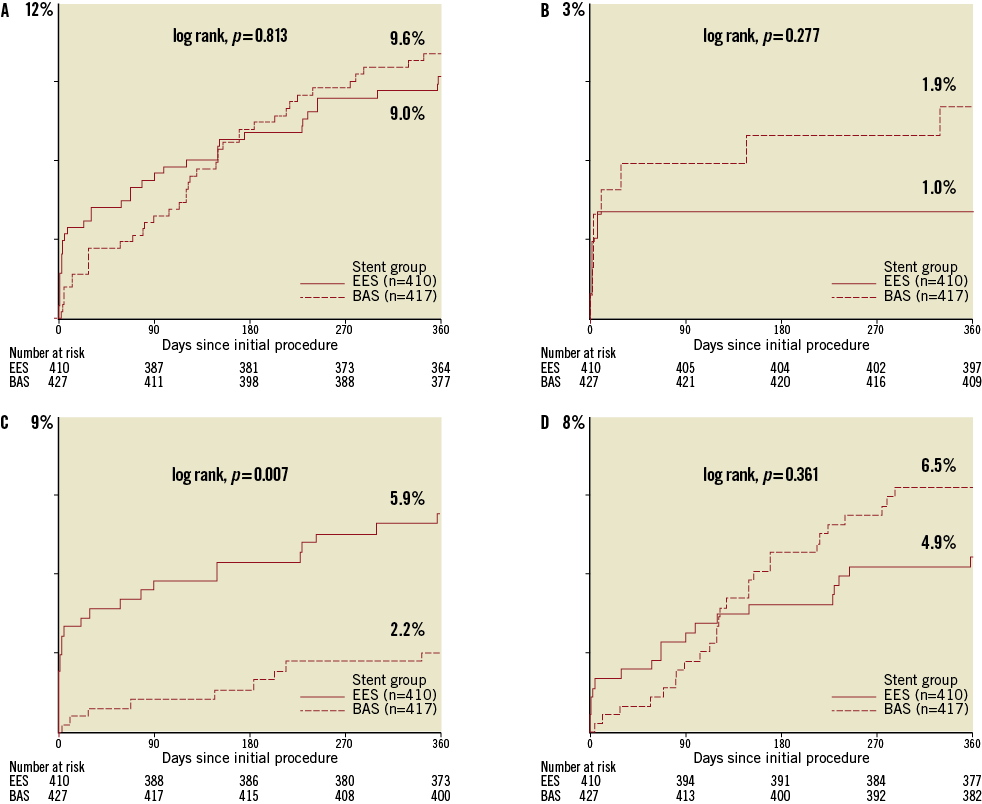
Figure 2. Kaplan-Meier estimates (cumulative incidence of events, %) for primary endpoints over 360 days of follow-up. Kaplan-Meier curves show the cumulative incidence of major adverse cardiac events (the primary endpoint), a composite of cardiac death, non-fatal myocardial infarction, or ischaemia-driven target lesion revascularisation (A); cardiac death (B); non-fatal myocardial infarction (C); and ischaemia-driven target lesion revascularisation (D). BAS: bioactive stent; EES: everolimus-eluting stent.
Among the individual components of the primary endpoint, non-fatal MI was less frequent in the BAS group as compared with the EES group (2.2% vs. 5.9%, respectively; p=0.007). Landmark analysis of the occurrence of MI during the first 30 days and from 30-day to 12-month follow-up is shown in Figure 3. During the first 30 days post-PCI, the rate of non-fatal MI was lower in the BAS group as compared with the EES group (0.7% vs. 3.2%, respectively; p=0.011). During the period from 30 days to 12 months, the cumulative occurrence of non-fatal MI was again lower in the BAS group (1.4% vs. 2.8%, respectively; p=0.019). However, the individual rates of cardiac death and ischaemia-driven TLR were similar between the two groups (1.9% vs. 1.0%, and 6.5% vs. 4.9%, respectively; p=0.39 and 0.37, respectively) (Figure 2).
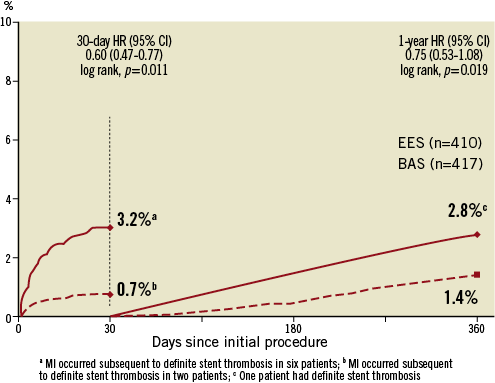
Figure 3. Landmark analyses of the occurrence of MI during the first 30 days and from 30-day to 12-month follow-up. BAS: bioactive stent; EES: everolimus-eluting stent
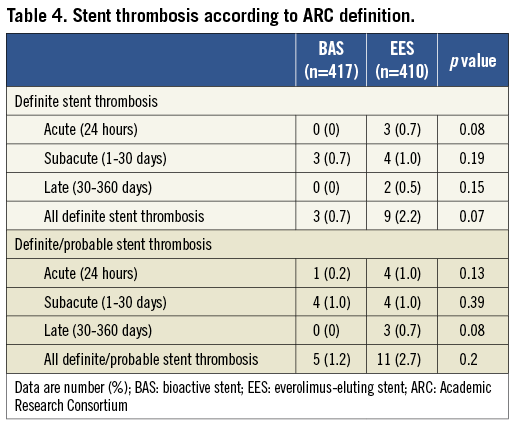
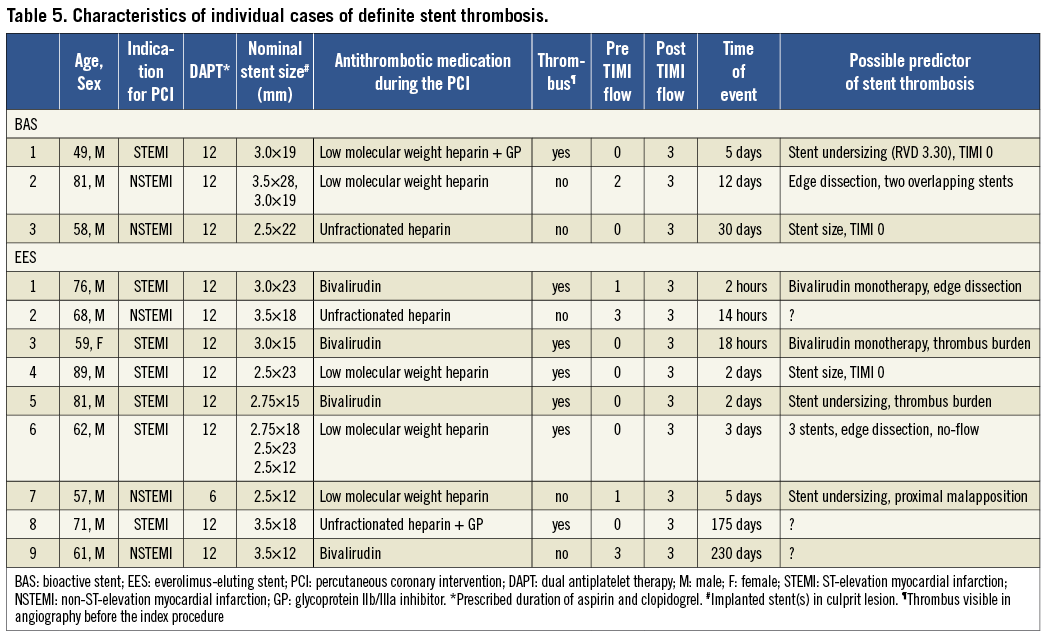
Among the secondary endpoints, the rates of all-cause death and the composite of cardiac death or non-fatal MI occurred at similar frequencies in the BAS group as compared with the EES group (p=0.42 and p =0.11, respectively). The rate of ARC-definite ST showed a trend to be less frequent in patients who received BAS as compared with those who received EES (0.7% vs. 2.2%, respectively; p =0.07) (Table 4). Characteristics of individual cases of ARC-definite ST are shown in Table 5. In the BAS group, there were three cases (0.7%) of subacute ST, whereas no cases of acute or late ST occurred in this group. In the EES group, there were three cases (0.7%) of acute ST, four cases (0.9%) of subacute ST, and two cases (0.5%) of late ST.
PREDICTORS OF ADVERSE CLINICAL OUTCOME
In univariate analyses, the predictors of MACE at 12-month follow-up were age, diabetes mellitus, hypertension, calcified lesions, the number of lesions treated, reference vessel diameter, and stent diameter. In multivariable analysis, independent predictors of MACE were age (OR 1.02, 95% CI 1.01-1.05, p =0.04), calcified lesions (OR 1.9, 95% CI 1.15-3.17, p=0.01), and the number of lesions treated (OR 1.54, 95% CI 1.05-2.26, p=0.03).
Discussion
MAJOR FINDINGS
The BASE-ACS trial reports a head-to-head randomised comparison of a BAS with EES in the setting of ACS. In patients undergoing early percutaneous coronary intervention for ACS, the insertion of BAS was non-inferior to EES with respect to the occurrence of the primary composite endpoint of MACE at 12-month follow-up. The relative risk ratio of MACE for BAS was 1.07 (0.6% absolute risk difference) when compared with EES, a difference that met the chief aim of the trial for non-inferiority of BAS in reducing MACE in this patient category. The trial was not adequately powered to address the individual components of safety and efficacy, but we did observe that non-fatal MI occurred less frequently, and ARC-definite ST trended to be lower in the BAS group as compared with the EES group.
EFFICACY AND SAFETY ENDPOINTS
Virtually all randomised controlled trials comparing DES with non-DES in patients with acute MI have demonstrated a significant reduction of re-intervention rates without an increase in the rates of death, recurrent MI, or ST19. A meta-analysis (n=9,470 patients) confirmed the benefit of DES with consistent findings of lower rates of revascularisation, and similar rates of adverse clinical events20. On the other hand, the 12-month clinical follow-up of BAS vs. paclitaxel-eluting TAXUS Liberté™ stents in patients presenting with acute MI demonstrated similar rates of efficacy and safety endpoints21.
Not surprisingly, there was a slight increase of ischaemia-driven TLR rates (efficacy endpoint) with BAS at 12-month follow-up, but this was compensated by a significant reduction of non-fatal MI (safety endpoint) with BAS at the same time point. The higher rate of non-fatal MI in the EES group is in accordance with the trend towards more ARC-definite ST in this group. Interestingly, whereas the time-to-event curves for TLR were initially in favour of BAS, the curves cross at approximately 4-month follow-up, and eventually diverge till the end of 12-month follow-up (Figure 2D). It is noteworthy that the lack of routine angiographic follow-up may have influenced the relative and absolute rates of TLR in the two groups; however, our clinically-driven protocol reflects real-world practice. Longer-term follow-up will determine whether these relative differences are durable over time.
Non-fatal MI events were more likely to occur with EES during the first two weeks following the index procedure. We cannot provide any definite explanation for this finding. The failed delivery of EES in four cases, especially with the slightly higher prevalence of left anterior descending artery as the target vessel might suggest a delivery issue with EES. This can be viewed in the light of the relatively high prevalence of calcified plaques in the EES group (41.2%). A hard calcified plaque offers far more resistance to stent expansion than a non-calcified one, and therefore carries a higher probability of incomplete stent strut apposition. The association between incomplete stent apposition and the occurrence of ST has been reported before. Furthermore, in an ad hoc study by Mosseri et al, more extensive coronary calcification (as revealed by intravascular ultrasound) was associated with periprocedural non-Q-wave MI22. Actually, in multivariable analyses, calcified lesion-type was an independent predictor of MACE. Although the prevalence of calcified lesions was similarly high in the BAS group (43.9%), the favourable mechanical properties of BAS may offer more adequate stent expansion than those offered by EES.
Moreover, the use of EES in more stable groups of patients was associated with lower rates of non-fatal MI and ARC-definite ST at one-year follow-up in the SPIRIT IV trial (1.9% and 0.3%, respectively), the RESOLUTE all-comers trial (4.1% and 0.3%, respectively), and the COMPARE trial (3.0% and 0.4%, respectively)23-25. Clearly, the enrolment of higher-risk patients with ACS would have played a key role in the higher incidence of non-fatal MI and definite ST in the EES group in the current trial (5.9% and 2.2%, respectively) at the same time point. Prior trials enrolled all-comer populations with a much lower risk profile than the current trial. In this regard, histopathological studies demonstrated that culprit sites in acute MI patients show a greater delay in arterial healing with evidence of persistent fibrin deposition and incomplete stent strut coverage as compared with culprit sites of patients presenting with stable angina who had underlying fibroatheromas and thick fibrous caps; the prevalence of late ST was also significantly higher26. Yet, recent data on “unrestricted” use of EES in routine clinical practice have shown that very late ST occurred at a steady rate of 0.2% per year at four-year follow-up27.
Early ST occurred in 0.7% of cases in the BAS group, as compared with 1.7% of cases in the EES group (Table 5). Initial TIMI flow grade 0 or 1 was observed in two out of three cases in the former, and in six out of seven cases in the latter group. Baseline TIMI flow grade 0/1 was among the independent predictors of early ST in a recent post hoc analysis of the HORIZONS-AMI trial28. Moreover, bivalirudin was used as the sole antithrombotic therapy during the procedure in two out of three cases of acute ST in the EES group. High rates of acute ST in association with periprocedural bivalirudin use were previously reported in patients with acute MI undergoing primary angioplasty in the HORIZONS-AMI trial29. Other possible underlying mechanisms of early ST include stent undersizing, high thrombus burden, or edge dissection. Interestingly, all cases of ST occurred while patients were on dual antiplatelet therapy (Table 5).
DUAL ANTIPLATELET THERAPY
In the current study, 99.3% of patients in the EES group were maintained on clopidogrel therapy at six months and 68.3% at 12 months (in the BAS group, 89.7% and 51.3%, respectively, p value for comparison with the EES group <0.001 for both). These figures are consistent with those reported from the SPIRIT III trial in which the proportion of patients receiving dual antiplatelet therapy at 12-month follow-up was 71.2% and 70.4%, for EES and paclitaxel-eluting stents, respectively9. Perhaps the substantial drop of patients in the EES group off the dual antiplatelet drug coverage during the period between six and 12-month follow-up has some association with the slowly rising time-to-event curves for non-fatal MI and ST for the EES group during this period of follow-up. Furthermore, despite a lower prevalence of patients maintained on dual antiplatelet therapy at 12-month follow-up, patients in the BAS group developed less non-fatal MI and ST at this point of follow-up.
STUDY LIMITATIONS
Although the current trial was well powered to detect non-inferiority of BAS as compared with EES concerning the primary composite endpoint of total MACE at 12-month follow-up, it was not adequately powered to address the individual components of safety and efficacy, such as cardiac death, non-fatal MI, or ischaemia-driven TLR, nor the secondary endpoints, such as all-cause death, the composite of cardiac death or non-fatal MI, and ARC-definite ST. Secondly, the unblinded nature of the trial is a weakness of the study design. Moreover, the current trial is not an all-comer trial; instead, some exclusion criteria existed on a background population presenting with ACS. Exclusion criteria such as aorto-ostial lesions and lesions longer than 28 mm might have, to some extent, favoured the outcome of BAS, introducing selection bias. Additionally, the failed delivery of EES in four cases (as compared with 0 in the BAS group) might suggest a delivery issue with EES. Furthermore, the enrolment of high-risk patients with ACS would account for the higher incidence of MI and ST in the EES group in the current trial as compared with those in previous trials23-25. We also acknowledge the limitation that dual antiplatelet therapy was not extended for 12 months, as recommended by the last update of guidelines for ACS. Whether the results of the current trial can be extrapolated to other second-generation DES would provide a potential avenue for future research. Finally, the TLR was clinically driven, and it may be that an angiographically-driven protocol might well have had a different outcome.
Conclusions
In the current prospective randomised BASE-ACS trial, among selected patients presenting with ACS who underwent early percutaneous coronary intervention and were followed without routine follow-up angiography, BAS achieved a rate of cumulative MACE at 12-month follow-up that was statistically non-inferior to EES.
Funding
This work was supported by grants from the Finnish Foundation for Cardiovascular Research, Helsinki, Finland. This work was also supported by unrestricted institutional grant from Hexacath, Paris, France; however, the company had no role in study design, data collection, data analysis and interpretation or manuscript writing.
Acknowledgements
This study was presented in late breaking clinical trials-session, EuroPCR Course, Paris, France, May 24, 2011. The authors thank Tuija Vasankari, RN, Eija Niemelä, RN, and Minna Ampio, RN, for their support in the conduct of this study. We gratefully acknowledge the help of the research nurses and medical staff in participating hospitals whose cooperation made this study possible.
Study sites and investigators: Heart Center, Satakunta Central Hospital, Pori, Finland (P P Karjalainen, A Ylitalo, J Mikkelsson, W Nammas); Department of Internal Medicine, Division of Cardiology, University of Oulu, Oulu, Finland (M Niemelä, K Kervinen, H Romppanen); Department of Medicine, Turku University Hospital, Turku, Finland (J KE Airaksinen, M Pietilä); Department of Cardiology, Kokkola Central Hospital, Kokkola, Finland (J Sia); Department of Medicine, Jyväskylä Central Hospital, Jyväskylä, Finland (K Nyman); CHU de Charleroi, Charleroi, Belgium (J Lalmand, P Lefebvre, A Aminian, D Dolatabadi); GHDC Charleroi, Charleroi, Belgium (M Carlier, S Fasseaux); Cardiovascular Center Aalst, OLV-Clinic, Aalst, Belgium (B De Bruyne); Department of Cardiology, Brighton and Sussex University Hospital NHS Trust, Brighton, UK (A de Belder); La Princesa University Hospital, Madrid, Spain (F Rivero-Crespo, J Salamanca-Viloria); Infanta Cristina University Hospital, Badajoz, Spain (JR Lopez-Minguez, JM Nogales Asensio, A Martinez Naharro, G Martinez Caceres); Swiss Cardiovascular Center, Bern University Hospital, Bern, Switzerland (O M Hess†); Department of Cardiology, Helsinki University Central Hospital, Helsinki, Finland (M Laine), Department of Cardiology and Vascular Medicine, Dr. Hasan Sadikin Hospital, Bandung, Indonesia (P Tedjokusumo).
Conflict of interest statement
The authors have no conflicts of interest to declare.
