Introduction
Among the 24 million people affected worldwide by mitral regurgitation1, almost two-thirds suffer from secondary mitral regurgitation (SMR)2. Moreover, the presence of symptomatic SMR in patients with heart failure (HF) remains a marker of increased mortality and rehospitalisation risk whatever its severity2. Thus, recent guidelines recommend downsized mitral annuloplasty as a standalone procedure (Class IIb) or as a concomitant procedure combined with coronary artery bypass grafting (CABG; Class I in European and IIa in American guidelines) for the management of SMR34. However, standard annuloplasty remains an intracardiac procedure requiring aortic cross-clamping and cardiopulmonary bypass and is thus associated with an increased risk of perioperative complications.
We aimed to evaluate the safety and efficacy of a new surgical procedure of extracardiac annuloplasty using the BACE (Basal Annuloplasty of the Cardia Externally) device (Phoenix Cardiac Devices) for the management of SMR in patients with systolic HF.
Methods
Forty-seven symptomatic patients with significant SMR (i.e., at least moderate or grade 2+) and systolic HF (i.e., left ventricular ejection fraction [LVEF] between 25% and 50%) referred for a surgical mitral valve (MV) intervention were prospectively recruited in 12 multinational centres between November 2012 and July 2019. Specific inclusion and exclusion criteria are described at ClinicalTrials.gov: NCT02701972. Patients had baseline, preoperative, 1-, 3-, 6-, 12-, and 24-month assessments including medical history, functional status (i.e., New York Heart Association [NYHA]), quality of life (i.e., Minnesota Living with Heart Failure Questionnaire [MLHFQ]) and echocardiography analysed by the independent echocardiography core laboratory based at the Quebec Heart & Lung Institute and following the American Society of Echocardiography standards and recommendations5. This study was conducted in conformity with the Declaration of Helsinki and Good Clinical Practice principles and approved by local ethics committees and respective health authorities. All patients provided informed written consent.
The primary safety endpoint was the freedom from major device- or surgery-related adverse events at 6 months following the procedure and was adjudicated by an independent Data Safety & Monitoring Board. The primary efficacy endpoint was the reduction of the SMR grade to ≤mild at 6 months. It was then evaluated in the subset (n=35) which had the 6-month follow-up (FU). Secondary endpoints were freedom from major device-related adverse events, and changes in the SMR grade, NYHA class and MLHFQ score between baseline and FU.
Results
Among the 47 patients recruited, implantation of the device was attempted but was not completed in 3 patients. Thus, procedural success was 94% (Figure 1). In the 44 patients (mean age±standard deviation [SD]: 62±12 years, 73% male, median [interquartile range] Charlson Comorbidity Index: 3 [2-4]) who were treated with the device, the majority (35 patients; 80%) underwent concomitant CABG, whereas 9 (20%) patients had a standalone procedure. The procedure was performed off-pump in 20 (57%) patients, and the median number of grafts was 3 (2–4).
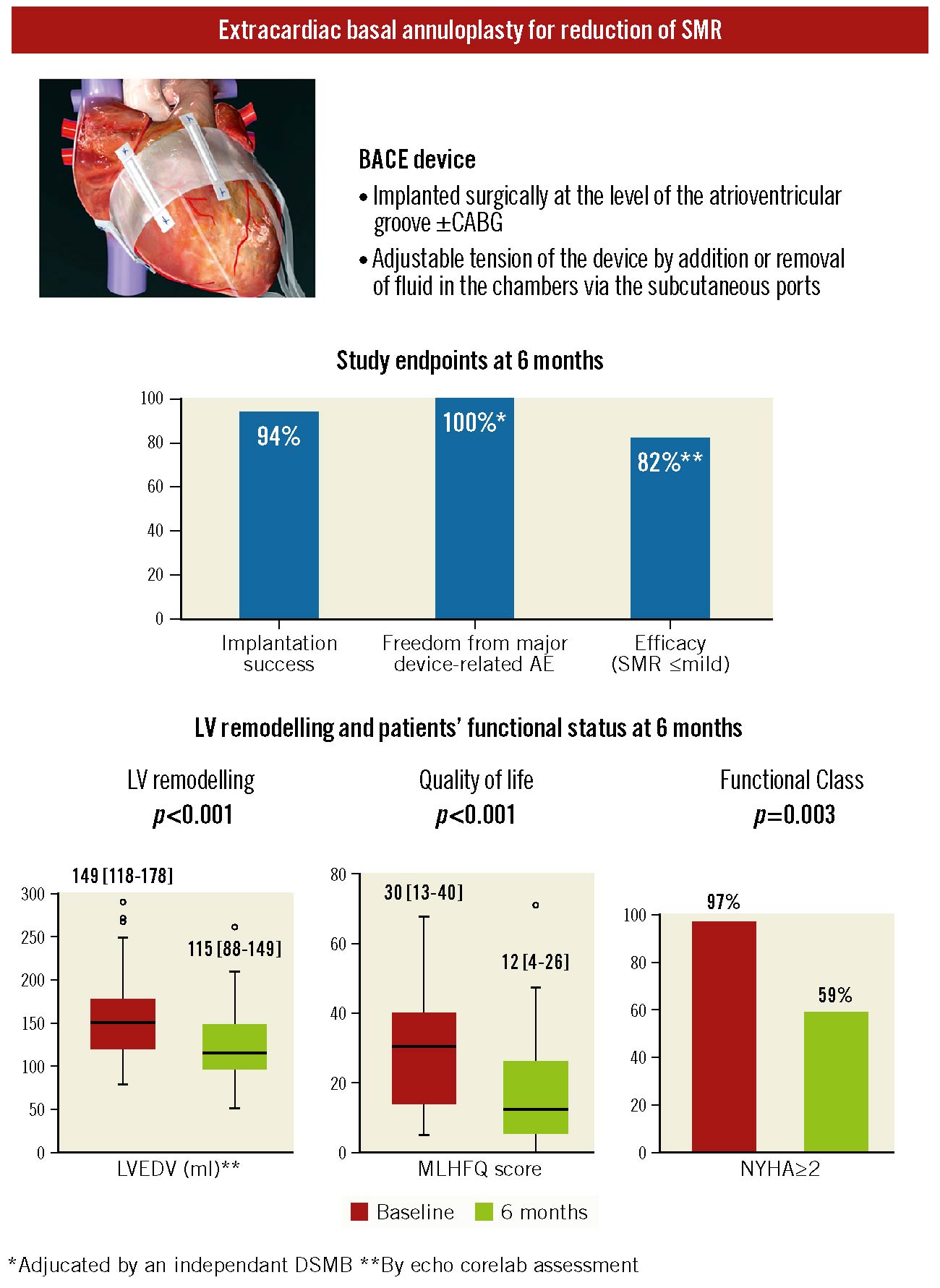
Figure 1.
Study endpoints and changes in mitral regurgitation severity, LV remodelling, quality of life and functional status at 6 months following the implantation of the BACE device. Illustration of the BACE device, which provides external annuloplasty of the cardiac base in order to reduce secondary mitral regurgitation severity and left ventricular dilation, and the main characteristics of the device (top); results of feasibility, safety, and efficacy endpoints (middle); and graphs for LV remodelling, NYHA Functional Class, and quality of life improvements (bottom) at 6 months after the implantation of the device in patients with at least moderate SMR and reduced ejection fraction. AE: adverse events; BACE: basal annuloplasty of the cardia externally; CABG: coronary artery bypass grafting; DSMB: Data Safety & Monitoring Board; LV: left ventricular; LVEDV: LV end-diastolic volume; MLHFQ: Minnesota Living with Heart Failure Questionnaire; NYHA: New York Heart Association; SMR: secondary mitral regurgitation
The primary safety endpoint was met in 70% of patients (31 of 44 patients implanted). Freedom from major device-related events was 100% and 98% at 6 and 12 months, respectively (i.e., compression of right ventricular inflow and outflow causing underfilling and reduced function in 1 patient who underwent surgical device removal). Two patients (5%) died before 30 days, and 6 patients died between 30 days and 6 months. None of these deaths were related to the device.
The primary efficacy endpoint was achieved in 82% of patients (SMR severity: 8 [24%] none or trace, 19 [58%] mild, 4 [12%] moderate and 2 [6%] severe; p-value vs baseline by Friedman test: p<0.001). In terms of secondary efficacy endpoints, 87% of patients had a reduction of at least one grade at 30 days, and 88% at 6 months. Echocardiographic parameters of left ventricular (LV) geometry/function and MV morphology (i.e., MV annulus diameter, tenting area, leaflet coaptation distance, indexed left atrial volume, LV end-diastolic volume, and LV end-systolic and end-diastolic diameters) significantly improved from baseline to 6 months (Figure 1) (all p-values by paired t-tests ≤0.003).
At 6 months, patients experienced significant improvement in NYHA class (p<0.001 by Friedman test), with percentages decreasing from 34% in Class III-IV to 12%, and from 63% in Class II to 47% (Figure 1). The MLHFQ score showed a marked improvement (i.e., reduction; paired t-tests: p<0.001) at 6 months (Figure 1). MR reduction (i.e., ≤mild) persists at 12 and 24 months, respectively, in 83% (20/24) and 91% (21/23) of patients who underwent these subsequent FU.
Discussion
The purpose of the BACE device is to achieve an extracardiac, indirect, and targeted restrictive mitral annuloplasty with the advantages of being less invasive than intracardiac downsized/restrictive annuloplasty and of offering the possibility of correcting persistent/recurrent MR by adjustment of the degree of annular restriction via the subcutaneous ports. Recurrent MR is believed to be the main reason for the lack of superiority of downsized annuloplasty versus mitral valve replacement for the treatment of ischaemic MR. The possibility of adjusting the degree of annular restriction achieved by the BACE device at any time during post-procedural follow-up may overcome this important limitation of standard intracardiac annuloplasty.
Limitations
The main limitations of this study are the single-arm design and the high prevalence of concomitant CABG procedures, which implicate that we cannot ascertain the respective effects of extracardiac annuloplasty versus myocardial revascularisation on study endpoints.
Conclusions
In summary, extracardiac basal annuloplasty with the BACE device appears to be safe and feasible and is associated with a significant reduction of SMR severity, positive LV remodelling, and improvement in the patient’s quality of life and functional status. Future randomised controlled trials comparing BACE±CABG versus intracardiac annuloplasty±CABG are needed.
Acknowledgements
We thank the participants of this study and the sponsor who was involved in the study design, the determination of performance goals and assisted with data interpretation, but who did not participate in data collection and data analysis.
Funding
This study was funded by Phoenix Cardiac Devices, Inc., Cary, NC, USA. J. Bernard is supported by a PhD research grant from the Canadian Institutes of Health Research (CIHR), Ottawa, Ontario, Canada. J. Beaudoin is supported by the Fonds de Recherche Québec - Santé, Montréal, Québec, Canada. P. Pibarot holds the Canada Research Chair in Valvular Heart Diseases and is supported by a Foundation grant (FDN-143225) from CIHR.
Conflict of interest statement
K. Talluri was the vice-president of clinical development at Phoenix Cardiac Devices. P. Pibarot received funding from Edwards Lifesciences, Medtronic and Phoenix Cardiac Devices for echocardiography core laboratory analyses, with no personal compensation. The other authors have no conflicts of interest to declare relevant to this study.
