Abstract
Cardiogenic shock (CS) is a devastating and fatal complication of acute myocardial infarction (AMI). CS can affect the pharmacokinetics and pharmacodynamics of medications. The unique properties of cangrelor make it the optimal P2Y12 inhibitor for CS-AMI, in terms of both efficacy and safety. The DAPT-SHOCK-AMI trial (ClinicalTrials.gov: NCT03551964; EudraCT: 2018-002161-19) will assess the benefits of cangrelor in patients with an initial CS-AMI undergoing primary angioplasty. This randomised, multicentre, placebo-controlled trial of approximately 550 patients (with an allowed 10% increase) in 5 countries using a double-blind design will compare initial P2Y12 inhibitor treatment strategies in patients with CS-AMI of (A) intravenous cangrelor and (B) ticagrelor administered as crushed tablets at a loading dose of 180 mg. The primary clinical endpoint is a composite of all-cause death, myocardial infarction (MI), or stroke within 30 days. The main secondary endpoints are (1) the net clinical endpoint, defined as death, MI, urgent revascularisation of the infarct-related artery, stroke, or major bleeding as defined by the Bleeding Academic Research Consortium criteria; (2) cardiovascular-related death, MI, urgent revascularisation, or heart failure; (3) heart failure; and (4) cardiovascular-related death, all (1-4) within 1 year after study enrolment. A platelet reactivity study that tests the laboratory antiplatelet benefits of cangrelor, when given in addition to standard antiplatelet therapy, will be conducted using vasodilator-stimulated phosphoprotein phosphorylation. The primary laboratory endpoints are the periprocedural rate of onset and the proportion of patients who achieve effective P2Y12 inhibition. The DAPT-SHOCK-AMI study is the first randomised trial to evaluate the benefits of cangrelor in patients with CS-AMI.
Introduction
MAGNITUDE OF THE ISSUE
The estimated global annual incidence of acute myocardial infarction (AMI) exceeds 7 million1. The average incidence of cardiogenic shock (CS) in patients hospitalised for AMI (CS-AMI) is approximately 7.5%2. Most CS-AMI cases have ST-segment elevation MI (STEMI)3. An analysis based on extensive US population data (from 1,000 hospitals) documented 44 CS-AMI cases per 100,000 hospitalisations in 2004 and 103 per 100,000 hospitalisations in 20183.
The incidence of CS developing during hospitalisation has been decreasing (currently 3.5%), whereas the incidence of initial (primary) shock has been stable or increasing (currently 4.6%)45. Furthermore, there has been an upward trend in preadmission cardiac arrests in patients with CS-AMI6.
PHARMACOTHERAPY-RELATED SPECIFICS
CS can affect all aspects of drug pharmacokinetics and pharmacodynamics78. The splanchnic circulation is hypoÂperfused because of reflexive vasoconstriction, which is further potentiated during treatment with vasopressor agents – the consequent ischaemia results in liver and kidney dysfunction. The preferred mode of drug administration is parenteral, and drugs that do not undergo metabolism are favoured.
In patients with CS-AMI, metabolism becomes less predictable, which can result in potentially serious adverse events due to overexposure or underexposure to active ingredients9. Antithrombotic medications are associated with a risk of bleeding; therefore, drugs with a rapid offset of action and antidotes are particularly beneficial.
EVIDENCE FOR SURVIVAL BENEFIT
Despite advances in medical treatment, reperfusion using primary percutaneous coronary intervention (PCI) remains the only intervention that improves the prognosis of patients with CS-AMI21011. An essential component of mechanical reperfusion in AMI is adjuvant antithrombotic pharmacotherapy, which is critical in preventing local thrombus progression and distant embolisation.
ANTIPLATELET THERAPY
Dual antiplatelet therapy using the newer oral P2Y12 receptor inhibitors (iP2Y12), i.e., ticagrelor or prasugrel, in combination with aspirin, is recommended in patients with AMI undergoing PCI based on the results of the TRITON-TIMI 38 (A Comparison of Prasugrel and Clopidogrel in Acute Coronary Syndrome Subjects Who Are to Undergo Percutaneous Coronary Intervention)12 and PLATO (A Comparison of Ticagrelor and Clopidogrel in Patients With Acute Coronary Syndrome)13 randomised trials. However, CS was an exclusion criterion in the TRITON study, and in the ticagrelor arm of the PLATO study, only 25 patients (0.7%) suffered from CS.
STUDY JUSTIFICATION
CS is the most common cause of death in patients with AMI who survive until hospital admission2. The in-hospital mortality rate of patients suffering from primary CS at admission is 44.4%5, and 20% of the deaths occur during the initial PCI procedure14. Characteristics including older age, multivessel coronary artery disease, increased time between symptom onset and reperfusion, and postprocedural Thrombolysis in Myocardial Infarction (TIMI) flow <3 were identified as independent predictors of death in patients with CS-AMI15. Rapid and effective platelet inhibition can modify two of the prognosis predictors in CS patients with the highest thrombotic risk, i.e., time until reperfusion and angioplasty outcomes (Figure 1).
The efficient inhibition of platelet aggregation is essential for preventing ischaemic events. The short-term risk of reinfarction is 3-4 times higher (9-12%)5 in patients with CS. Additionally, CS is the strongest independent predictor of stent thrombosis16. Ischaemic stroke occurs in 2.4% of patients with CS; this number rises for more invasive circulatory stabilisation methods17.
Patients with CS-AMI are also at risk of severe bleeding. Typically, bleeding occurs in 1 out of 5 CS-AMI cases during early hospitalisation18.
Cangrelor is the best studied iP2Y12 with a parenteral mode of application (Table 1). The effective inhibition of adenosine diphosphate (ADP)-induced platelet aggregation occurs 2 minutes after initiating treatment, and the antiplatelet effect is maintained throughout the infusion period19. Cangrelor metabolism is independent of splanchnic organ function and does not affect liver enzyme-metabolised drugs. Platelet aggregation is restored approximately 60 minutes after stopping the administration of the drug19.
Cangrelor therapy initiated concomitantly with crushed ticagrelor tablets in patients with STEMI undergoing primary PCI results in prompt and potent platelet inhibition during the intervention; additionally, cangrelor therapy bridges the gap until the full antiplatelet efficacy of ticagrelor is achieved20.
The unique pharmacokinetic and pharmacodynamic properties of cangrelor make it the optimal iP2Y12 for CS-AMI in terms of efficacy and safety. The degree to which iP2Y12 suppresses ADP-mediated platelet function depends on the potency of the antiplatelet drug and the baseline (before treatment) prothrombotic condition; this is most pronounced in STEMI patients. A better understanding of the relationship between study medication-related platelet reactivity inhibition (through quantification of the rate of onset and intensity of inhibition during the peri-PCI period) and clinical outcomes in one trial may contribute to developing more effective and safer treatment strategies21.
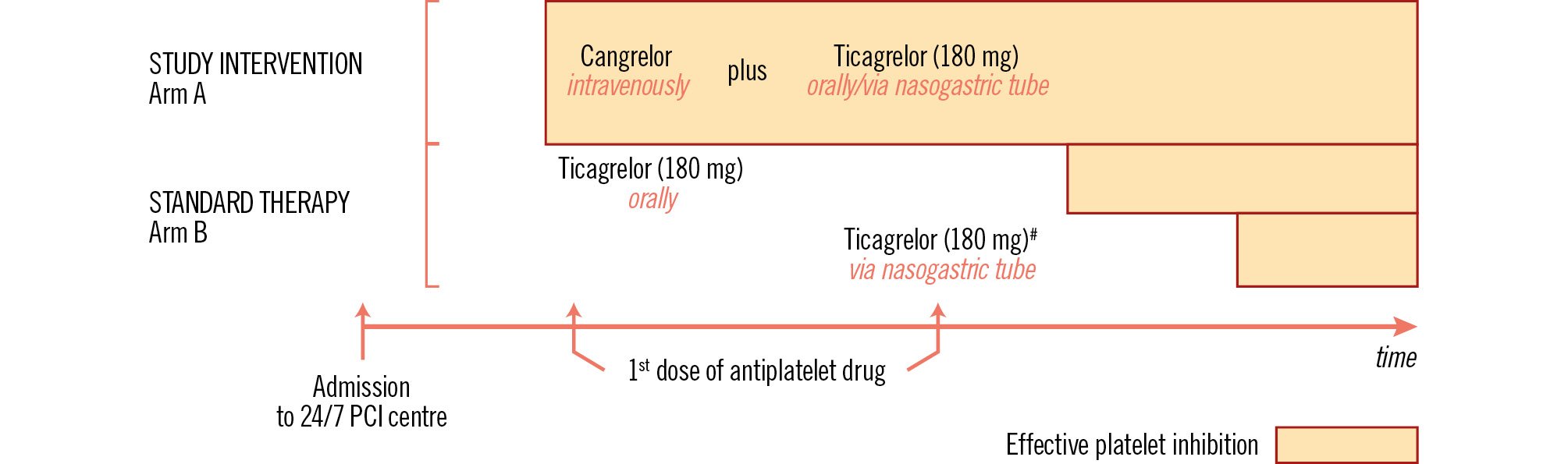
Figure 1. Benefits of the tested study therapy (study rationale). The first arrow indicates CS-AMI patients’ admission to a 24/7 PCI centre, usually directly to the catheterisation laboratory. The upper part of the figure shows patients randomised to cangrelor IV (arm A) achieving effective platelet inhibition immediately after initiation of therapy (large orange bar), irrespective of conscious state and oral intake ability. The lower part shows patients assigned to arm B and treated with ticagrelor. Patients who are able to take ticagrelor orally (second arrow) achieve effective platelet inhibition after hours (medium orange bar). Patients with impaired consciousness cannot receive ticagrelor until after introducing the nasogastric tube (often after arriving at the catheterisation laboratory; third arrow), which represents a further delay in the onset of ticagrelor antiplatelet efficacy (small orange bar). The onset of effective inhibition of platelet reactivity (expressed by colour fill) depends on the antiplatelet drug administered. #Patients with impaired consciousness. CS-AMI: cardiogenic shock complicated acute myocardial infarction; IV: intravenous; PCI: percutaneous coronary intervention
Table 1. P2Y12 receptor inhibitors.
| Drug | Structure | Effect | Reversibility | Method of use | Frequency of use |
|---|---|---|---|---|---|
| Ticlopidine | Thienopyridine | Indirect | No | Oral | BID |
| Clopidogrel | Thienopyridine | Indirect | No | Oral | QD |
| Prasugrel | Thienopyridine | Indirect | No | Oral | QD |
| Ticagrelor | ATP analogue | Direct | Yes(half-life 6-12 hr) | Oral | BID |
| Cangrelor | ATP analogue | Direct | Yes(half-life 3 min) | Parenteral | Continuousinfusion |
| ATP: adenosine triphosphate; BID: twice a day; QD: once a day | |||||
Study design
STUDY OBJECTIVES
The Dual Antiplatelet Therapy For Shock Patients With Acute Myocardial Infarction trial (DAPT-SHOCK-AMI; ClinicalTrials.gov: NCT03551964, Protocol numbers: 13062017-23-1, EudraCT: 2018-002161-19) is a double-blind, multicentre, international, placebo-controlled trial testing the hypothesis that intravenous cangrelor is (a) more effective in terms of its rate of onset and the proportion of patients achieving effective periprocedural inhibition of ADP-induced platelet aggregation and (b) at least as effective as the recommended treatment of oral (crushed) ticagrelor in reducing major cardiovascular events in patients with initial CS-AMI indicated for primary PCI strategy.
STUDY POPULATION
The study population will be comprised of patients who meet the inclusion criteria, defined as follows: (1) over 18 years of age; (2) AMI according to the ESC/ACC/AHA definition22 with an indication for emergency PCI (primary PCI strategy); (3) CS due to an AMI present upon admission meeting at least 2 of the following criteria23: (a) systolic blood pressure <90 mmHg in the absence of hypovolaemia, (b) need for vasopressor and/or inotropic therapy, and (c) signs of organ hypoperfusion (cyanosis, cold extremities, disorders of consciousness, or heart failure); (4) signed informed consent form as per the applicable legal regulations and regulatory authority requirements; and (5) women with childbearing potential should avoid pregnancy and use a highly effective method of contraception throughout the study period (relevant for long-term use of ticagrelor).
The exclusion criteria are presented in Supplementary Table 1.
RANDOMISATION
The patients are randomised in a 1:1 ratio using random permuted blocks, stratified by study centre. Randomisation is performed using an interactive web-response system developed by the Institute of Biostatistics and Analyses, Faculty of Medicine, Masaryk University, Czech Republic on the base of the TrialDB system.
STUDY MEDICATION
The patients are randomised into one of two treatment arms (Figure 2) according to the study intervention: arm A: cangrelor versus arm B: ticagrelor.
Patients in arm A will receive the active study medication, cangrelor, as an intravenous (IV) bolus of 30 μg/kg (application <1 min) followed immediately by a continuous infusion at 4 μg/kg. Thirty minutes before the end of the cangrelor infusion, 180 mg of ticagrelor (crushed tablets) will be administered, followed by a maintenance dose of 90 mg every 12 hours (Supplementary Figure 1)2425.
In arm B of the study, the patients will receive 180 mg of crushed ticagrelor tablets orally as the loading dose, and thereafter a maintenance dose of 90 mg twice daily, as per the guidelines. The placebo dosage, forms, and methods of administration (cangrelor-placebo and ticagrelor-placebo) are identical to those of their respective active substance (cangrelor and ticagrelor). Thus, the cangrelor- and ticagrelor-placebo treatments will be administered in the same way as the IV cangrelor and oral ticagrelor (as crushed tablets), respectively.
Randomisation and initiation of the study medication administration of both compared study arms should be performed immediately (at the earliest possible time) after the patient’s admission to the 24/7 PCI centre, which is usually the catheterisation laboratory.
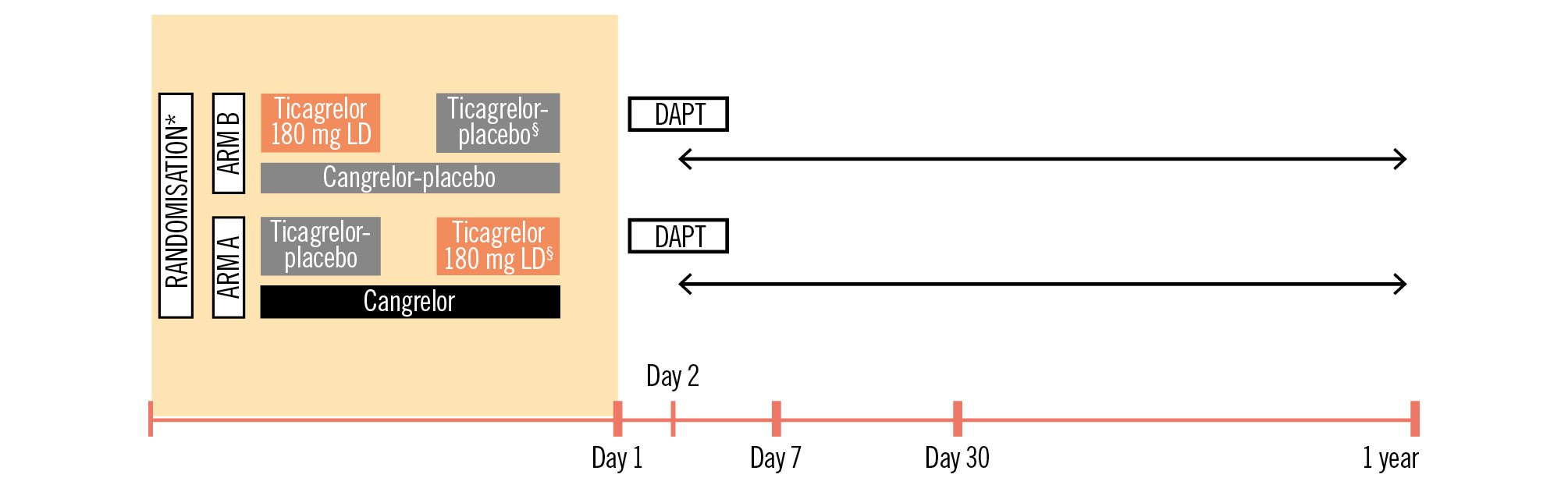
Figure 2. Study design. Study initiation should be as soon as possible after admission to the hospital. *Study medication administered in addition to initial aspirin IV; §administered 30 minutes before the end of the infusion. Visits: day 7 after randomisation, day 30±5 days, 1 year±14 days. Ticagrelor was in crushed form. DAPT consists of a P2Y12 inhibitor plus aspirin. DAPT: dual antiplatelet therapy; IV: intravenous; LD: loading dose
CONCOMITANT THERAPY
The antiplatelet therapy used in this study is iP2Y12, which will be administered in addition to an initial aspirin dose of 500 mg IV, followed by 100 mg of aspirin as a daily oral dose. Proton pump inhibitors are recommended to prevent gastrointestinal bleeding. The administration of other standard-care therapies, including additional adjuvant antithrombotic therapy (e.g., a bailout glycoprotein [GP] IIb/IIIa inhibitor and parenteral antithrombin drugs) and mechanical circulatory support, will be left to the discretion of the attending physician.
STUDY FOLLOW-UP
Patients enrolled in the study will be followed for 12 months. A summary of the timing of the visits and examinations that will be performed is presented in Table 2. Recommendations regarding treatment during the follow-up period, including dual antiplatelet therapy, will adhere to the appropriate guidelines.
Table 2. Scheduled visits during the 1-year study follow-up.
|
Randomisation |
Day 7 |
Day 30±5 days Visit 3 |
Year 1±14 days Visit 4 |
|
|---|---|---|---|---|
| Clinical condition | X | X | X | X |
| ECG | X | X | X | X |
| Echocardiography | X | X | X | X |
| #MRI32 | - | X | X | X |
| §¶Laboratory sampling | §¶X | ¶X | ¶X | ¶X |
| Questionnaire on quality of life (EuroQol 5D)33 | - | - | X | X |
| #MRI substudy – in selected centres. Laboratory examination involves the following: §examination of the effectiveness of antiplatelet therapy by the determination of VASP phosphorylation via flow cytometry – in selected centres; ¶haematological and biochemical blood tests. ECG: electrocardiogram; MRI: magnetic resonance imaging; VASP: vasodilator-stimulated phosphoprotein |
||||
CLINICAL ENDPOINTS
The primary endpoint is defined as a composite of death, myocardial infarction, or stroke 30 days after enrolment into the study. The secondary endpoints are summarised in Supplementary Table 2. Other goals include conducting a cost-effectiveness analysis and a magnetic resonance imaging substudy of the predefined endpoints.
PLATELET REACTIVITY STUDY
Patients who meet the enrolment criteria and are randomised at the 5 selected centres will be eligible for the platelet reactivity study. The laboratory antiplatelet effectiveness of the cangrelor and ticagrelor loading dose-based initial iP2Y12 strategies will be determined by vasodilator-stimulated phosphoprotein (VASP) phosphorylation using flow cytometry, which is the most specific method for verifying and quantifying the effectiveness of iP2Y12 and is associated with clinical outcomes. The determinations will be performed by an accredited facility using standardised sampling kits and protocols specified by the manufacturer. The design of the laboratory study is illustrated in Figure 3. The tests will be carried out as follows: before initiating treatment with the antiplatelet study drugs, upon completion of the coronary intervention procedure, 1 hr after PCI, 2 hrs after PCI, at the end of the cangrelor infusion, 1 hr after the end of the infusion, and 2 hrs after the end of the infusion. The primary laboratory endpoint will be assessed at the second and third VASP examinations. Monitoring platelet function dynamics after the intervention will provide important insights into the study − testing a strategy of combined intravenous and oral treatment with P2Y12 inhibitors.
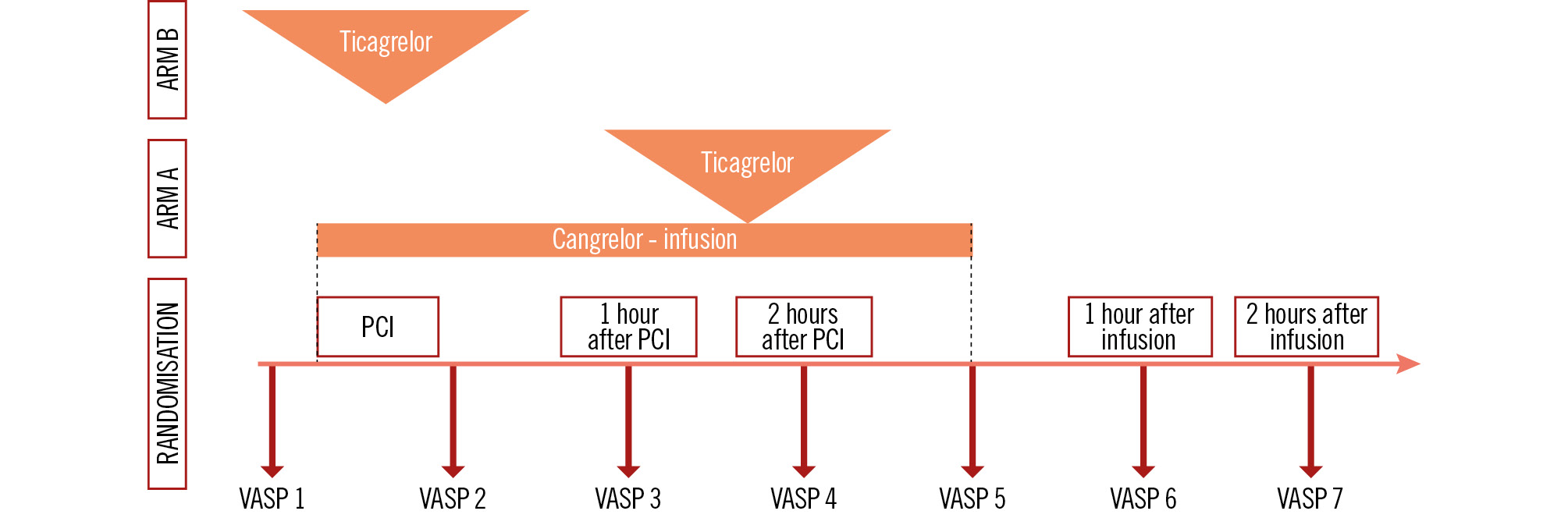
Figure 3. Platelet reactivity study design. Timing of VASP evaluations: VASP 1 – baseline (before study treatment administration), VASP 2 – at the end of the PCI procedure, VASP 3 – 1 hour after PCI, VASP 4 – 2 hours after PCI, VASP 5 – at the end of the cangrelor infusion, VASP 6 – 1 hour after the end of the infusion, and VASP 7 – 2 hours after the end of the infusion. PCI: percutaneous coronary intervention; VASP: vasodilator-stimulated phosphoprotein
SAMPLE SIZE
This study was initially designed to include 304 patients. However, since the beginning of the study, there has been a substantial amount of new evidence to consider when calculating study population sizes. The power analysis was computed for the superiority and non-inferiority scenarios under the assumption of primary endpoint occurrences in previous studies and registries, as presented in Supplementary Table 3.
The null hypothesis for the primary clinical endpoint was the equality of event rates, and the alternative hypothesis was the inequality of event rates between the analysed groups. Based on an expected event rate of 50% in the control group versus 38% in the cangrelor group, a required power of 80%, and a 2-sided statistical significance level of 5%, 536 patients would be needed to detect a 12% difference between groups and reject the null hypothesis. Allowing for a 3% dropout rate, 550 patients should be enrolled in the study (with a permitted 10% increase). The dropout rate is based on several clinical studies.
Based on an expected event rate of 50% in the control group versus 40% in the cangrelor group (difference 10%), a non-inferiority margin of 1%, a required power of 80%, and a 2-sided statistical significance level of 5%, 506 patients would be required to accept the additional non-inferiority hypothesis.
A power analysis for the platelet reactivity study, with the endpoint of effective inhibition of the platelet VASP <50%, was computed for superiority, requiring a power of 80% and a 2-sided statistical significance level of 5%. The null hypothesis for this endpoint was the equality of event rates, whereas, for the alternative hypothesis, it was the inequality of event rates between the analysed groups. Based on an expected event rate of 70% in the control group versus 90% in the cangrelor group, 124 patients will be required to detect a 20% difference between groups and reject the null hypothesis. The required sample size falls within the practical limits of VASP measurements, which is approximately 150.
The power analysis was computed using the PASS 13 software (2014 [NCSS, LLC]).
STATISTICAL ANALYSES
Statistical analyses will be performed using the SPSS Statistics software, version 28.0.1.1 (IBM). The analyses will be performed using an intention-to-treat principle supplemented by a modified intention-to-treat principle, which only includes patients who receive a dose of the study drug. A sensitivity analysis according to the per-protocol population will also be performed.
Standard descriptive statistics will be calculated in the analysis, i.e., absolute and relative frequencies for categorical variables, and medians supplemented with 5th-95th percentiles or means supplemented with standard deviations for continuous variables. The chi-square test will be used to test the statistical significance of differences in the primary endpoint and all other categorical variables, and the Mann-Whitney U test will be used for continuous variables.
Univariate and multivariate logistic regression or Cox proportional hazards models will be used as additional analyses of the influence of patient characteristics on the endpoint occurrence, and the Kaplan-Meier methodology will be adopted to visualise time-to-event data.
The level of statistical significance will be set at p=0.05. All the statistical analyses will be performed according to the U.S. Food and Drug Administration Guidance Document “E9 Statistical Principles for Clinical Trials” (FDA-1997-D-0508).
STUDY ORGANISATION
This study is a non-commercial, investigator-initiated study, and it is an international project that will be implemented by teams of investigators in 5 countries (the Czech Republic, France, Germany, Poland, and the Slovak Republic). It adheres to the ethical principles of the Declaration of Helsinki, the International Conference on Harmonization of Good Clinical Practice (R2), and all other applicable legal and regulatory requirements, including the General Data Protection Regulation. The study’s organisational structure includes executive, steering, and endpoint adjudication committees, and a data safety monitoring board. External clinical research associate organisations will monitor all entries in the electronic case report forms and the completeness of the source documentation.
The institutions’ ethics committees will conduct yearly audits of trial protocols and progress during the study. The auditing process is independent of both trial investigators and trial sponsors.
The records of the procedural findings, coronary angiograms, and PCIs will be submitted to the coordinating centre, where they will be evaluated by an independent panel of experts blinded to how medication was allocated within the study.
The assessment of VASP phosphorylation, as part of the platelet reactivity study, will be performed by an external laboratory and personnel blinded to the allocation of the study medication. The laboratory will enter the results directly into a database that will be inaccessible to the investigators.
Discussion
The increasing average age of the global population and the rising incidence of coronary heart disease indicate that the number of people at risk of CS-AMI is growing. However, conducting randomised studies to assess treatments in patients with CS is challenging. Circulatory instability is usually an exclusion criterion for participation in clinical trials attempting to verify the benefits of antithrombotic pharmacotherapies12. The currently available evidence is limited to that from small studies and registry data (Supplementary Table 3).
Minimising thrombotic risk and restoring coronary blood flow at the microcirculatory level is critical for reperfusion and a better prognosis for CS-AMI26. The safety and efficacy of adjuvant combination antiplatelet therapy is mainly determined by the selection of the optimal inhibitor for ADP-induced platelet activation to be used alongside aspirin. Adding a third antiplatelet drug, such as a GP IIb/IIIa inhibitor, to the combination with aspirin and especially the highly effective iP2Y12, ticagrelor, increases the risk of significant bleeding, thereby nullifying any potential benefits in terms of patient outcomes27. Cangrelor, in addition to all the other advantages already mentioned for patients with CS-AMI, reduces the periprocedural need for bailout rescue GP IIb/IIIa inhibitors28.
The initial P2Y12 inhibitor treatment strategy with intravenous cangrelor is compared to the crushed tablet form of ticagrelor. This form of ticagrelor loading dose demonstrated a faster onset of effective platelet inhibition than the oral dose (whole tablet)29. Therefore, despite the lack of evidence of the benefit on patient prognosis, crushed ticagrelor is recommended as the preferred mode of administration in patients with CS2630.
The VASP method was selected to monitor the rate of onset and extent of inhibition of P2Y12 platelet receptors by the compared drugs. This choice was based on the unique specificity of the VASP assay for the P2Y12 signalling pathway, which makes it the only method for monitoring P2Y12 inhibitor efficacy that is not influenced by the P2Y1 receptor functional status31. Moreover, unlike other assays, such as the widely used point-of-care VerifyNow (Werfen), VASP phosphorylation measurements are not affected by the co-administration of a GP IIb/IIIa inhibitor31, which is frequently used during primary angioplasty for CS-AMI.
Conclusions
The DAPT-SHOCK-AMI study aims to provide clinical evidence for selecting the appropriate antiplatelet therapies in patients with AMI complicated by initial CS undergoing primary PCI and, thus, potentially improve the prognosis of this often fatal condition (Figure 4).
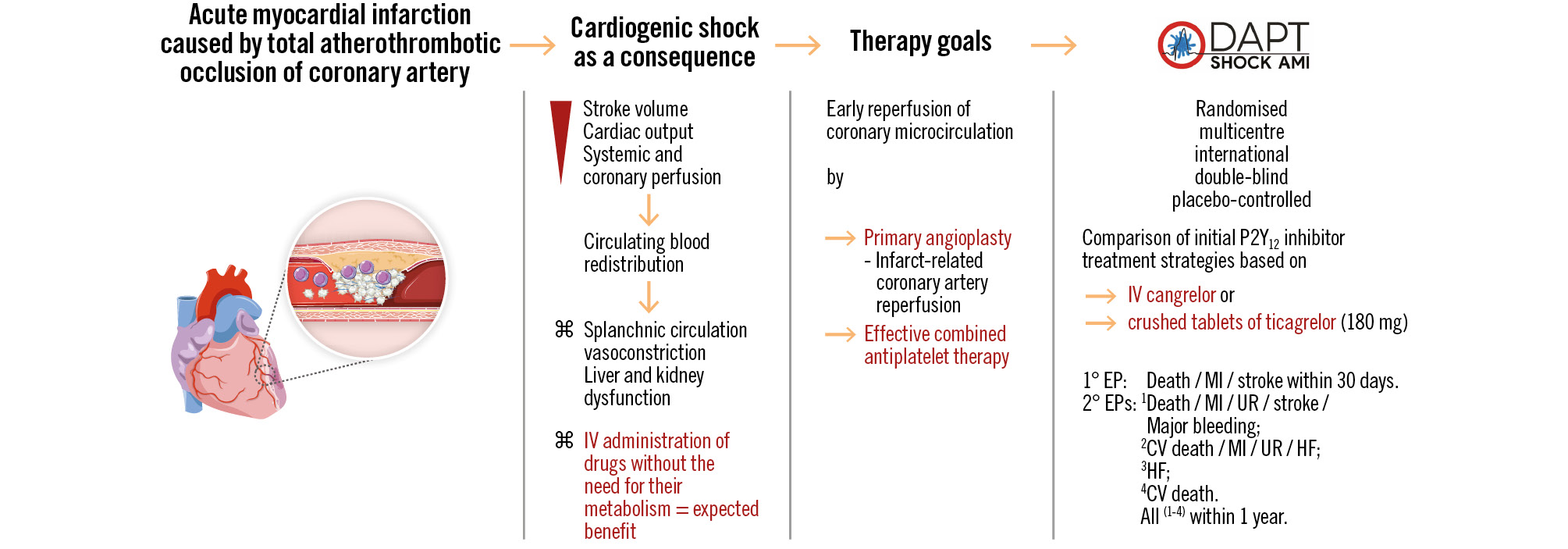
Figure 4. Study endpoints. The figure summarises the factual basis for conducting the study and outlines the study outcome endpoints. ⌘ refers to the associated data. CV: cardiovascular; EP: endpoint; HF: heart failure; IV: intravenous; MI: myocardial infarction; UR: urgent revascularisation
Acknowledgements
We wish to acknowledge and recognise all those who have participated in the preparation of this academic project and all the researchers, study nurses, and coordinators who will participate in its implementation, especially Katerina Pilikova (study coordinator for the Czech Republic and Slovak Republic), Denisa Odvodyova (head study nurse for the Czech Republic and Slovak Republic), Veronika Dantlingerova, Bc. (coordination of the international part of the study). The authors express their special thanks to Pharm Dr Lukas Laznicka from University Hospital Kralovske Vinohrady in Prague (Czech Republic) and Pharm Dr Jan Tomasch from VULM Bratislava (Slovak Republic) for their professional help in the field of pharmacotherapy. The authors also acknowledge and thank Dr Zuzana Grycova, MSc, Irena Babilona, MSc, Terezia Kuricova, MSc, and Sarka Matyskova from CZECRIN for their help in ensuring the study’s international implementation. Expressions of appreciation and thanks are also due to the teams involved in the setup of the study in Germany: Mrs Timea Keller, Mrs Isabela Kast (Centre for Clinical Trials, University Hospital Tübingen); France: Mrs Karine Brouchard (ACTION group); and Poland: Mrs Jolanta Dwojacka and Dr Pawel Dyras (Brillance Sp z o.o.).
Funding
Costs associated with the implementation of the project will be covered by the Ministry of Health of the Czech Republic (Grant No. NV19-02-00086 [principal investigator, ZM]). All rights are reserved. This work is further supported by the Charles University Czech Republic Research Programs, PROGRESS Q 38 and COOPERATIO - Cardiovascular Science, Research Support from Donatio Universitatis Carolinae, awarded to ZM (principal investigator of the study), and the Charles University 4EU+ mini-grant (No. 4EU+/23/F1/04). The study is also supported by the project National Institute for Research of Metabolic and Cardiovascular Diseases (Program EXCELES, ID Project No. LX22NPO5104) - funded by the European Union - Next Generation EU.
Conflict of interest statement
Z. Motovska discloses the following relationships: research funding related to the implementation of the DAPT-SHOCK-AMI study: the Ministry of Health of the Czech Republic Grant No. NV19-02-00086; the Charles University Czech Republic: research programmes PROGRESS Q 38 and COOPERATIO - Cardiovascular Science, research support from Donatio Universitatis Carolinae, the Charles University 4EU+ mini-grant (No. 4EU+/23/F1/04); Programme EXCELES, ID Project No. LX22NPO5104 - funded by the European Union - Next Generation EU; advisory boards: AstraZeneca, Bayer, Boehringer Ingelheim, Chiesi, Sanofi; research funds for the institution or honoraria from: AstraZeneca, Amgen, Bayer, Czech Society of Cardiology, Idorsia, Janssen, RITA-MI 2 Grant agreement ID: 899991, HORIZON 2020-EU 3.1.1; meeting attendance support: Czech Society of Cardiology, Boehringer Ingelheim, Pfizer. D.L. Bhatt discloses the following relationships - advisory board: Angiowave, Bayer, Boehringer Ingelheim, CellProthera, Cereno Scientific, Elsevier Practice Update Cardiology, High Enroll, Janssen, Level Ex, McKinsey, Medscape Cardiology, Merck, MyoKardia, NirvaMed, Novo Nordisk, PhaseBio, PLx Pharma, Stasys; board of directors: American Heart Association New York City, Angiowave (stock options), Bristol-Myers Squibb (stock), DRS.LINQ (stock options), High Enroll (stock); consultant: Broadview Ventures, GlaxoSmithKline, Hims, SFJ, Youngene; data monitoring committees: Acesion Pharma, Assistance Publique-Hôpitaux de Paris, Baim Institute for Clinical Research (formerly Harvard Clinical Research Institute, for the PORTICO trial, funded by St. Jude Medical, now Abbott), Boston Scientific (chair, PEITHO trial), Cleveland Clinic, Contego Medical (chair, PERFORMANCE 2), Duke Clinical Research Institute, Mayo Clinic, Mount Sinai School of Medicine (for the ENVISAGE trial, funded by Daiichi Sankyo; for the ABILITY-DM trial, funded by Concept Medical; for ALLAY-HF, funded by Alleviant Medical), Novartis, Population Health Research Institute, Rutgers University (for the NIH-funded MINT trial); honoraria: American College of Cardiology (Senior Associate Editor, Clinical Trials and News, ACC.org; Chair, ACC Accreditation Oversight Committee), Arnold and Porter law firm (work related to Sanofi/Bristol-Myers Squibb clopidogrel litigation), Baim Institute for Clinical Research (formerly Harvard Clinical Research Institute; RE-DUAL PCI clinical trial steering committee funded by Boehringer Ingelheim; AEGIS-II executive committee funded by CSL Behring), Belvoir Publications (Editor-in-Chief, Harvard Heart Letter), Canadian Medical and Surgical Knowledge Translation Research Group (clinical trial steering committees), CSL Behring (AHA lecture), Cowen and Company, Duke Clinical Research Institute (clinical trial steering committees, including for the PRONOUNCE trial, funded by Ferring Pharmaceuticals), HMP Global (Editor-in-Chief, Journal of Invasive Cardiology), Journal of the American College of Cardiology (Guest Editor; Associate Editor), K2P (Co-Chair, interdisciplinary curriculum), Level Ex, Medtelligence/ReachMD (CME steering committees), MJH Life Sciences, Oakstone CME (Course Director, comprehensive review of interventional cardiology), Piper Sandler, Population Health Research Institute (for the COMPASS operations committee, publications committee, steering committee, and USA national co-leader, funded by Bayer), WebMD (CME steering committees), Wiley (steering committee); other: Clinical Cardiology (Deputy Editor); patent: Sotagliflozin (named on a patent for sotagliflozin assigned to Brigham and Women’s Hospital who assigned to Lexicon; neither he nor Brigham and Women’s Hospital receive any income from this patent); research funding: Abbott, Acesion Pharma, Afimmune, Aker Biomarine, Alnylam, Amarin, Amgen, AstraZeneca, Bayer, Beren, Boehringer Ingelheim, Boston Scientific, Bristol-Myers Squibb, Cardax, CellProthera, Cereno Scientific, Chiesi, CinCor, Cleerly, CSL Behring, Eisai, Ethicon, Faraday Pharmaceuticals, Ferring Pharmaceuticals, Forest Laboratories, Fractyl, Garmin, HLS Therapeutics, Idorsia, Ironwood, Ischemix, Janssen, Javelin, Lexicon, Lilly, Medtronic, Merck, Moderna, MyoKardia, NirvaMed, Novartis, Novo Nordisk, Otsuka, Owkin, Pfizer, PhaseBio, PLx Pharma, Recardio, Regeneron, Reid Hoffman Foundation, Roche, Sanofi, Stasys, Synaptic, The Medicines Company, Youngene, 89Bio; royalties: Elsevier (Editor, Braunwald’s Heart Disease); site co-investigator: Abbott, Biotronik, Boston Scientific, CSI, Endotronix, St. Jude Medical (now Abbott), Philips, SpectraWAVE, Svelte, Vascular Solutions; trustee: American College of Cardiology; unfunded research: FlowCo. P. Kala discloses the following relationships - advisory board: Boston Scientific, Edwards Lifesciences, Chiesi, Novartis, Sanofi, Servier; speaker and consultancy fees: Abbott, Boston Scientific, Servier, Zentiva. T. Giesler discloses the following relationships: speaker and consultancy fees from AstraZeneca, Bayer, Bristol-Myers Squibb/Pfizer, Ferrer/Chiesi, Novartis, Medtronic, and Edwards Lifesciences; research grants from Bayer, Bristol-Myers Squibb/Pfizer, Ferrer/Chiesi, Medtronic, and Edwards Lifesciences. J. Belohlavek discloses consulting fees from Abiomed and Resuscitec; honoraria from Novartis, Boehringer Ingelheim, and AstraZeneca. G. Montalescot declares research funds for the institution or honoraria from: Abbott, Amgen, AstraZeneca, Ascendia, Bayer, BMS, Boehringer Ingelheim, Boston Scientific, Celecor, CSL Behring, Idorsia, Lilly, Medpace, Novartis, Novo Nordisk, Opalia, Pfizer, Sanofi, Terumo. J. Matejka declares speaker honoraria from AstraZeneca and Servier. C.B. Olivier reports research support from Deutsche Forschungsgemeinschaft, Deutsche Herzstiftung, Freiburg University, Else Kröner-Fresenius Stiftung, and Haemonetics; honoraria: Bayer Vital GmbH, BMS, Boehringer Ingelheim, Daiichi Sankyo, Ferrer, Idorsia, and Janssen. P. Ostadal discloses the following relationships - advisory board: AstraZeneca, Boehringer Ingelheim, Bristol-Myers Squibb, Sanofi; speaker and consultancy fees: Abiomed, Amgen, AstraZeneca, Bayer, Boehringer Ingelheim, Edwards Lifesciences, Fresenius, Getinge, Novartis, Pfizer, Sanofi, Servier. G. Ducrocq discloses speaker and/or consulting fees from Abbott, Amarin, Amgen, AstraZeneca, Bayer, Boston Scientific, BMS, Novo Nordisk, Sanofi, CEC, DSMB; steering committee in Amgen, Novo Nordisk, Janssen; proctoring for Boston Scientific; travel fees from Sanofi; ownership interest in Bioquantis. M. Karpisek discloses: co-investigator of BioVendor, BioLab Assays. P. Tomasov discloses travel fees from Boston Scientific and Euromedical. J. Kubica discloses lecture honoraria from AstraZeneca and Ferrer. The other authors have no conflicts of interest to declare.
Supplementary data
To read the full content of this article, please download the PDF.
