
Abstract
Aims: Our aim was to assess the clinical outcomes of the Direct Flow Medical Transcatheter Aortic Valve System (DFM-TAVS), when used in routine clinical practice.
Methods and results: This is a prospective, open-label, multicentre, post-market registry of patients treated with DFM-TAVS according to approved commercial indications. Echocardiographic and angiographic data were evaluated by an independent core laboratory and adverse events were adjudicated and classified according to VARC-2 criteria by an independent clinical events committee. The primary endpoint was freedom from all-cause mortality at 30 days post procedure. Secondary endpoints included procedural, early safety and efficacy endpoints at 30 days. Two hundred and fifty patients with severe aortic stenosis undergoing transcatheter aortic valve implantation (TAVI) with the DFM-TAVS were enrolled in 21 European centres. The primary endpoint, freedom from all-cause mortality at 30 days, was met in 98% (245/250) of patients. Device success was 83.8%. Moderate or severe aortic regurgitation was reported in 3% of patients, and none/trace regurgitation in 73% of patients. Post-procedural permanent pacemaker implantation was performed in 30 patients (12.0%).
Conclusions: The DFM-TAVS was associated with good short-term outcomes in this real-world registry. The low pacemaker and aortic regurgitation rates confirm the advantages of this next-generation transcatheter heart valve (THV).
Introduction
Transcatheter aortic valve implantation (TAVI) is a well-established, percutaneous treatment option for severe aortic stenosis patients with comorbidities that result in a prohibitive surgical risk profile1,2. The first-generation TAVI systems allowed only limited manoeuvring during valve positioning, and operators were unable to perform valve repositioning or retrieve the valve if the final result was suboptimal3,4. Suboptimal valve positioning is related to significant residual aortic regurgitation (AR), which is one of the most important predictors of clinical outcomes after TAVI5-7.
The pre-CE mark experience (DISCOVER trial) with the Direct Flow Medical® Transcatheter Aortic Valve System (DFM-TAVS) (Direct Flow Medical Inc., Santa Rosa, CA, USA) showed promising results, characterised by high rates of procedural efficacy and patient safety8. The DFM-TAVS, with its non-metallic, inflatable and deflatable structure, allows precise positioning, repositioning, assessment of valve performance in its final position, and, if required, retrieval. The implantation technique has been standardised9 and the DFM-TAVS is currently part of routine clinical practice in an increasing number of European centres. The aim of the present study was to evaluate procedural and clinical outcomes of the DFM-TAVS, as used in routine clinical practice.
Methods
PATIENT POPULATION AND ENDPOINT DEFINITIONS
This is an ongoing prospective open-label, single-arm, multicentre, post-market registry (ClinicalTrials.gov Identifier: NCT01845285) designed to evaluate clinical outcomes of the DFM-TAVS when used in routine clinical practice according to the approved commercial indications. The patient population included candidates for TAVI who were >70 years old, with severe, symptomatic aortic valve stenosis, and identified as extreme-risk candidates for open surgical aortic valve repair. The site investigators (interventional cardiologist and cardiothoracic surgeon) agreed that medical factors precluded surgery, based on the conclusion that the probability of death or serious morbidity exceeded the probability of meaningful improvement due to the patient’s comorbidities (such as, but not limited to, severe chronic obstructive pulmonary disease [COPD], porcelain aorta, previous thoracic irradiation) or logistic EuroSCORE ≥20. Here we report 30-day outcomes of the first 250 patients enrolled into the registry.
The primary endpoint was freedom from all-cause mortality at 30 days post procedure. Adverse events were reviewed by an independent clinical events committee (Appendix 1). Secondary endpoints were assessed at the time of procedure, and at 30 days. Safety endpoints were evaluated according to VARC-210. AR at follow-up was determined from transthoracic echo and graded according to VARC-2. When echocardiographic images at follow-up (discharge, 30 days) were available for core lab analysis (MedStar Health Research Institute, Washington DC, USA), haemodynamic core lab measurements (effective orifice area, mean transaortic pressure gradient, aortic regurgitation) superseded site-reported values. Haemodynamic determinants of procedural success (aortic regurgitation, mean transvalvular pressure gradient and aortic peak velocity) were established from the first reported data point after valve implantation. Clinical and haemodynamic outcomes (NYHA class, aortic regurgitation, mean transvalvular pressure gradient, effective orifice area) are presented for DFM-implanted patients only. Aortic regurgitation up to 30 days was established from the last available echocardiographic measurement within the 30-day follow-up period.
All potential study subjects underwent a pre-interventional screening process to determine eligibility. Informed consent was obtained prior to screening. Valvular anatomy was assessed using multi-slice computed tomography (MSCT) of the thoracic aorta. Annular diameters were determined from MSCT measurements of the annular perimeter. Vascular access was evaluated using MSCT of the abdominal aorta and iliofemoral vessels. Severe aortic valve stenosis was defined by echocardiographic criteria including a mean gradient >40 mmHg or peak jet velocity >4.0 m/s and aortic valve area ≤0.8 cm2 or aortic valve area index ≤0.5 cm2/m2.
DEVICE AND PROCEDURE DESCRIPTION
The DFM prosthesis has been described elsewhere11. In brief, it consists of a non-metallic, percutaneous, bovine pericardial valve with an inflatable Dacron polyester double-ring design containing a circuit of non-compliant balloons (Figure 1). The upper (aortic) and lower (ventricular) rings, interconnected by a system of pressurisable channels, can be pressurised independently through positioning wires, which are also designed to manoeuvre the valve during implantation. At the time of this study, four valve sizes were available (23/25/27/29 mm valve sizes for annular diameters of 19-22/21-24/24-26 /26-28 mm, respectively).
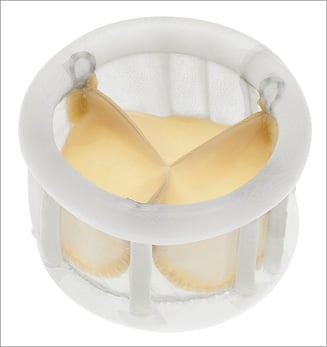
Figure 1. The Direct Flow Medical transcatheter aortic valve prosthesis.
After balloon valvuloplasty, the DFM delivery catheter is positioned in the left ventricle. The two rings are pressurised up to 12 atm by injecting a mixture of saline and contrast media through the positioning wires. After deflation of the aortic ring, the operator retracts and/or advances the three positioning wires, ensuring ventricular ring alignment to the native aortic annulus, followed by full aortic ring pressurisation. After assessment of the valve position, the rings can be depressurised and the prosthesis repositioned or completely retrieved, if necessary. In the latter case, both rings are deflated and the valve is pulled into a nitinol basket in the abdominal aorta and retrieved through the introducer sheath. When an optimal position has been obtained, the contrast-saline mixture is exchanged for a polymer while maintaining the pressure in the bioprosthesis at 12 atm. Once the polymer solidifies, the device is no longer retrievable8,11.
STATISTICAL ANALYSIS
Data are presented through summary statistics where categorical variables are summarised as frequencies and percentages and continuous variables through mean and standard deviation. Data were analysed with SAS version 9.4 software (SAS Institute Inc., Cary, NC, USA).
All available data have been included for the data sets as described in the tables and figures. Unless otherwise described, safety evaluations are based on all enrolled subjects, and efficacy measurements (NYHA class, gradient, EOA, AR) are based on the subjects implanted with the study valve. Changes from baseline for EOA and mean gradient were tested for statistical significance at each follow-up time point (discharge and 30 days) in a generalised linear mixed model with the corresponding baseline measurement and follow-up time as a fixed effects and subject as a random effect.
Results
PATIENT POPULATION AND PROCEDURAL CHARACTERISTICS
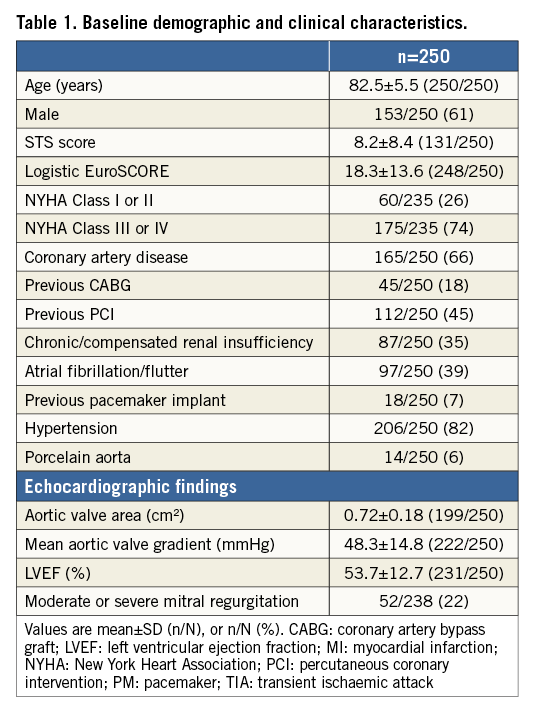
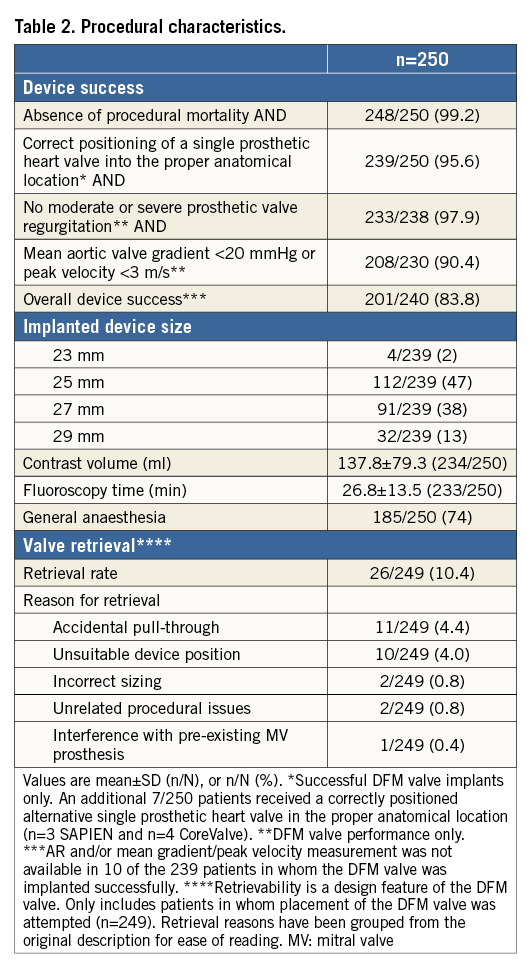
Two hundred and fifty patients with severe aortic stenosis undergoing TAVI with the DFM-TAVS were enrolled in 21 European centres (Appendix Figure 1). Follow-up at 30 days was available for all patients; 172 of 250 patients also had a medical assessment at 30 days following the index procedure, while the remainder had a telephonic follow-up. Baseline demographic, clinical and echocardiographic characteristics of the total population are shown in Table 1. The majority of the enrolled patients were in an advanced NYHA functional class (NYHA Class III-IV in 74%), while the logistic EuroSCORE was 18.3±13.6%. Procedural characteristics are shown in Table 2.
STUDY ENDPOINTS
The primary endpoint (freedom from all-cause mortality at 30 days) outcome was 98.0% (245/250 patients) (Table 3).
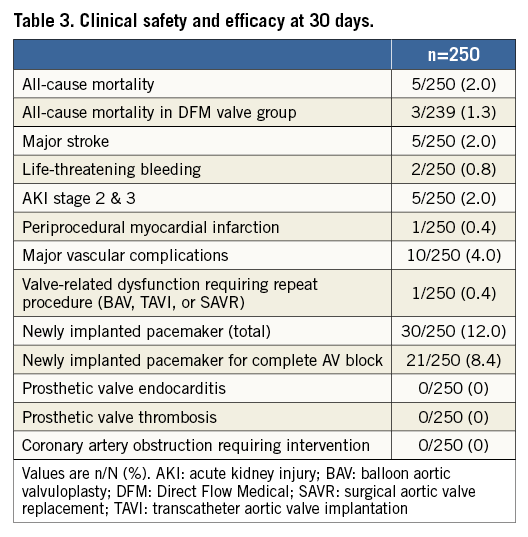
The Direct Flow Medical valve was successfully implanted in 239/250 patients, with no moderate or severe aortic regurgitation in 233/238 patients (Table 2). The retrievability feature of the device was successfully used in 10.4% of the procedures (the reasons for retrieval are reported in Table 2). Freedom from all-cause mortality was 98.0%, and 98.7% in patients who received a DFM valve (Table 3). Freedom from mortality and major stroke at 30 days was 96.4%. There were no cases of valve thrombosis, endocarditis or coronary obstruction requiring intervention. The 11 patients who were enrolled but did not receive a DFM-TAVS included one patient who died on day 0 due to annulus rupture after valvuloplasty, one patient who was converted to emergency surgery and successful implantation of a bioprosthesis after perforation of the left ventricle by pacemaker lead and cardiac tamponade prior to DFM valve placement, two patients who received BAV with successful retrieval of the DFM valve (due to persistent high gradient in one patient, and anatomy too long for the catheter system in another), and seven patients converted to other TAVI devices. Three patients were converted to SAPIEN valves (Edwards Lifesciences, Irvine, CA, USA) after successful retrieval of the DFM valve (valve anatomies did not allow an optimal placement of the DFM valve). Four patients were converted successfully to CoreValve® (Medtronic, Minneapolis, MN, USA) after retrieval of the DFM valve, when positioning of the DFM valve was unsuccessful; one of these four died on day 16 after multiple complications due to pre-existing low left ventricular ejection fraction.
New York Heart Association (NYHA) functional class was significantly improved at 30-day follow-up compared to baseline (Figure 2). Of note, the rate of pacemaker implantation for all 250 patients was 12% (30 patients); in 21 patients (8.4%) a complete atrioventricular block after TAVI necessitated pacemaker implantation. Residual moderate or severe AR at 30 days was observed in 3% of the patient population treated with the DFM valve (Figure 3). None/trace aortic regurgitation was observed in 73% of patients. The effective orifice area (EOA) improved significantly from 0.7 cm2 at baseline to 1.5 cm2 at 30 days. The mean transaortic pressure gradient decreased from 48.0 mmHg at baseline to 15.8 mmHg at 30 days (Figure 4).
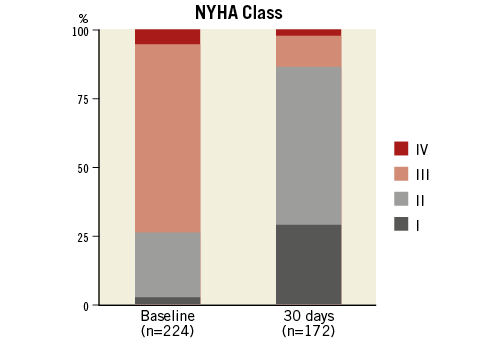
Figure 2. Percentage of patients in NYHA functional Class I-IV at baseline and at 30 days in patients implanted with the DFM valve. NYHA: New York Heart Association
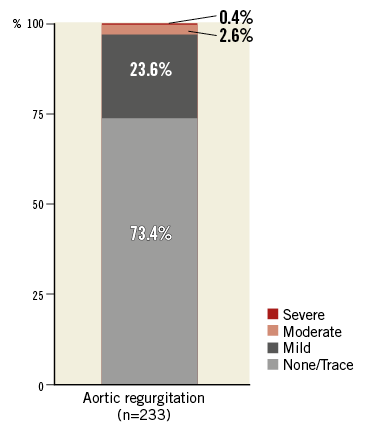
Figure 3. Aortic regurgitation up to 30 days according to echocardiographic VARC criteria. Aortic regurgitation was evaluated by the core lab in 93/233 patients at baseline, in 17 patients at discharge and in 76 patients at 30 days, and is reported by study sites in 140/233 patients at baseline, in 34 patients at discharge and in 106 patients at 30 days. VARC: Valve Academic Research Consortium
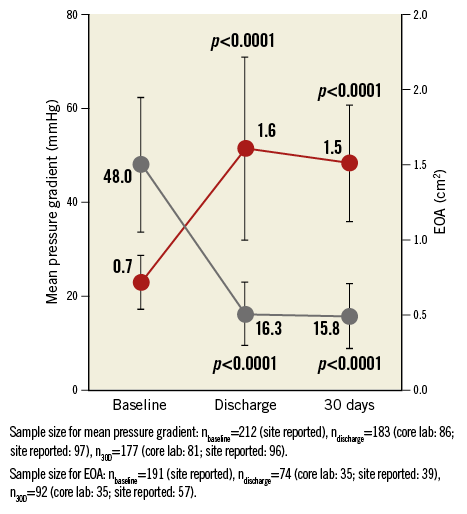
Figure 4. Effective orifice area and mean transaortic pressure gradient derived from transthoracic echocardiography at baseline, discharge and 30 days. P-values refer to difference over baseline. 30D: 30 days
Discussion
The main findings of the current study are the following. DFM-TAVS used in routine clinical practice resulted in a 98% freedom from all-cause mortality at 30 days (primary endpoint). Procedural success, early safety and efficacy measures compared favourably to similar real-world experiences. Residual AR at 30 days was low at 3%.
Primary and secondary endpoints in the real-world registry were similar to those reported for the pre-market DISCOVER trial8, even though inclusion and exclusion criteria were less strict in the current registry. For example, patients with left ventricular ejection fraction <30% or chronic kidney disease (creatinine >3.0 mg/dl), who would have been excluded from the DISCOVER trial, were enrolled in the registry patient cohort. Some factors that may have contributed to the consistently high efficacy and safety rates, despite a more challenging patient population, are the experience level of the high-volume centres and the standardisation of the DFM-TAVS positioning and deployment technique.
Earlier studies of first-generation devices reported higher mortality rates. The FRANCE-2 prospective registry reported a 30-day mortality rate of 8.5%12, and investigators of the GARY registry reported a 30-day mortality rate of 5.6% for the transfemoral approach patient cohort13. The current registry of the second-generation TAVI system from Direct Flow Medical reports a much lower 30-day all-cause mortality rate of 2.0%. A similar improvement has been noted for the Edwards SAPIEN 3 valve (Edwards Lifesciences)14 and the Lotus™ valve (Boston Scientific, Marlborough, MA, USA)15 (reporting 2.1% and 4.2%, respectively). This reflects the evolution in newly developed TAVI systems.
Another difference between first- and second-generation devices is observed in the degree of residual aortic regurgitation. A meta-analysis by Généreux et al of 16 studies of first-generation devices reported a residual moderate-to-severe AR of 7.4%16, whereas the rate of moderate-to-severe residual AR in the present study is only 3%. This compares favourably with recent experiences with the balloon-expandable Edwards SAPIEN 3, reporting an AR rate of 3.5% at 30 days14. Also, of note, the EOAs reported here and in the Edwards SAPIEN 3 study are identical at 1.5 cm2.
The rate of post-procedural permanent pacemaker implantation was lower (12.0%) than the previously described DISCOVER trial experience (17%)8. This difference may have resulted from the increased use of the standardised “inner curve” technique and the performance of only a single balloon valvuloplasty in the majority of cases, which leads to a higher implant position9. The pacemaker rate is also lower than that previously reported for the Medtronic CoreValve (24-29%) and similar to the recently reported rate for the Edwards SAPIEN 3 (13.3%)14,17. The REPRISE II study15 of another repositionable TAVS (Lotus valve) reports a post-procedural pacemaker implantation rate of 28.6%, which is considerably higher than the pacemaker rate reported here. The authors of the REPRISE II study attributed this relatively high rate to overstretching of the left ventricular outflow tract (LVOT) by the nitinol frame of the Lotus valve; in contrast, the DFM-TAVS has no metallic backbone and features an adjustable design.
The retrieval rate in this large patient cohort was 10.4%. In 0.8% of patients the valve was exchanged for a different size. This finding highlights the potential to improve device success by using the retrievability/repositionability feature. This feature is particularly useful in cases where valve sizing is difficult, or for the borderline in-between valve size cases, and may be important for eliminating post-procedural AR.
We report a general anaesthesia rate of 74%, depending on different practice applied in the different participating centres. Webb et al report an overall rate of 76.7% with the Edwards SAPIEN 3 prosthesis (63.5% for transfemoral use)14. All patients enrolled in the PARTNER trial (100%) underwent general anaesthesia18. The investigators of the FRANCE-2 registry reported a general anaesthesia rate of 59.2%12. A recent meta-analysis of Mayr et al reported a conversion rate to general anaesthesia of 17%, primarily due to vascular complications, and urgent intubation associated with haemodynamic stability19. These data suggest that the rate of TAVI patients undergoing general anaesthesia is variable in different clinical realities due to the lack of large-scale randomised studies to address this issue.
Study limitations
The echocardiographic core lab data evaluation is incomplete, because systematic collection of echo images was not emphasised at the outset of the study. We report a device success rate of 83.8% based on VARC-2 criteria. This finding was mainly driven by 9.6% of patients with a mean aortic valve gradient >20 mmHg or peak velocity >3 m/s. Meredith et al recently reported a 5.7% rate of peak velocity >3 m/s following Lotus valve implantation15. However, previous studies carried out with the DFM device also reported lower rates of transvalvular aortic gradient, varying from 1.9% to 7%8,20. These findings reflect the inherent limitation of the lack of a systematic core lab evaluation of the aortic valve gradient in the present real-world experience. In addition, only short-term (30-day) follow-up data are presented and findings could differ in long-term follow-up. Also, peak-to-peak gradient data were not systematically collected. Furthermore, differences in patient selection, risk stratification and procedural practices among the participating centres and operators cannot be excluded.
Conclusions
In this prospective, open-label, multicentre, post-market registry, the DFM-TAVS, as used in routine clinical practice, was effective and safe in the short-term follow-up, with low rates of mortality, stroke, vascular complications, and aortic regurgitation. The findings support further investigation of the DFM-TAVS in high- and intermediate-risk patients.
| Impact on daily practice We present our extensive, multicentre experience with the Direct Flow Medical Transcatheter Aortic Valve System (DFM-TAVS) for the treatment of patients with severe aortic stenosis. This second-generation TAVI system showed favourable clinical outcome at 30 days of follow-up. The retrievability/repositionability feature of the prosthesis proved to be useful in cases of difficulty in sizing and may have led to the relatively low aortic regurgitation rate observed in our patient population. |
Conflict of interest statement
H. Ince, P. Frambach, S. Kische, F. Gatto, U. Hink, W. Schillinger, S. Scholtz and P. den Heijer receive proctorship fees from Direct Flow Medical. A. Colombo is a minor shareholder in Direct Flow Medical. C. Naber and J. Schofer are consultants to Direct Flow Medical. A. Latib and F. De Marco are consultants to and proctors for Direct Flow Medical. F. Asch’s institution (MedStar Health Research Institute) receives fees from Direct Flow Medical for his work as echo core lab director. The other authors have no conflicts of interest to declare.
SUPPLEMENTARY DATA
Appendix 1. Study management
CLINICAL EVENTS COMMITTEE
Pierfrancesco Agostoni, MD, PhD, University Medical Center Utrecht
Giuseppe Biondi-Zoccai, MD, Department of Medico-Surgical Sciences and Biotechnologies, Universitá degli Studi di Roma La Sapienza
Giuseppe Bruschi, MD, Department of Cardiology & Cardiac Surgery, A.O. Ospedale Niguarda Ca’ Granda
Andrea Pascotto, MD, Ospedale dell’Angelo
Andrea Rocco, PhD, MSc, Charité Universitätsmedzin Berlin
Prof. Dr. med. Michael Schmoeckel, ASKLEPIOS Klinik St. Georg
STATISTICAL ANALYSIS
Tami Crabtree, MS, Senior Biostatistician, Consultant to DFM
Appendix Figure 1. Participating centres.
