Abstract
Percutaneous coronary intervention (PCI) is frequently performed for stable angina. However, the first blinded trial, ORBITA, did not show a placebo-controlled increment in exercise time in patients with single-vessel disease, at 6 weeks, on maximal antianginal therapy. ORBITA-2 will assess the placebo-controlled efficacy of PCI on angina frequency in patients with single- or multivessel disease, at 12 weeks, on no antianginal therapy. ORBITA-2 is a double-blind placebo-controlled trial randomising participants with (i) angina at presentation, (ii) documented angina during the 2-week pre-randomisation symptom assessment phase, (iii) objective evidence of ischaemia, (iv) single- or multivessel disease, and (v) clinical eligibility for PCI. At enrolment, antianginals will be stopped, and angina questionnaires completed. Participants will record their symptoms on a smartphone application daily throughout the trial and will undergo exercise treadmill testing and stress echocardiography at pre-randomisation. They will then undergo coronary angiography with unblinded invasive physiology assessment. Eligible participants will then be sedated to a deep level of conscious sedation and randomised 1:1 between PCI and placebo. After the 12-week blinded follow-up period, they will return for questionnaires, exercise testing and stress echocardiography assessment. If angina becomes intolerable, antianginals will be introduced using a prespecified medication protocol. The primary outcome is an angina symptom score using an ordinal clinical outcome scale for angina. Secondary outcomes include exercise treadmill time, angina frequency, angina severity and quality of life. Trial registration: ClinicalTrials.gov: NCT03742050
Introduction
More than 500,000 percutaneous coronary intervention (PCI) procedures are performed annually worldwide for stable coronary artery disease (CAD)1. The results of over 14,000 patients randomised to an invasive versus a conservative strategy, followed up for 4.5 years, showed no evidence of net reduction in mortality2345. These procedures are principally performed to relieve angina.
Unblinded randomised controlled trials (RCT) have consistently shown angina relief from PCI4678. The first blinded RCT, ORBITA (Objective Randomised Blinded Investigation with optimal medical Therapy of Angioplasty in stable angina), showed a surprisingly small placebo-controlled effect of PCI9.
However, ORBITA may not have provided the definitive picture for several reasons. First, it only enrolled patients with single-vessel disease, to allow the effect size to be later regressed10 against the severity of the lesion. Second, ORBITA participants received maximally tolerated antianginal medication to test the guideline-directed incremental effect of PCI on a background of antianginal therapy. This may have attenuated the potential benefit of PCI. Third, patients were enrolled based on clinically indicated PCI, with 94-96%1011 of patients having evidence of ischaemia before randomisation. Fourth, while patients needed to have symptoms prior to enrolment, there was no additional requirement to have angina episodes immediately before randomisation. 88% were in Canadian Cardiovascular Society (CCS) Class I to III at randomisation. This proportion was 89% in the FAME 2 (Fractional Flow Reserve versus Angiography for Multivessel Evaluation 2) and 88% in the COURAGE (Clinical Outcomes Utilizing Revascularization and Aggressive Drug Evaluation) trials312. Fifth, it prespecified change in treadmill exercise time, rather than symptoms, as the primary endpoint, similar to the unblinded ACME (Angioplasty Compared to Medicine) trial of balloon angioplasty6 and to mirror US Food and Drug Administration and the European Medicines Agency requirements for trials of antianginal therapy. Sixth, it selected a 6-week follow-up period, being long enough for resolution of ischaemia and short enough to be ethical and practical for the first placebo-controlled trial of PCI. Finally, ORBITA prespecified paired t-test methodology, as used by the positive unblinded ACME trial. This was not the most powerful statistical method for detecting treatment effect.
ORBITA-2 will provide the next test of the placebo-controlled efficacy of PCI in reducing angina, differing from ORBITA by parameters shown in Table 1.
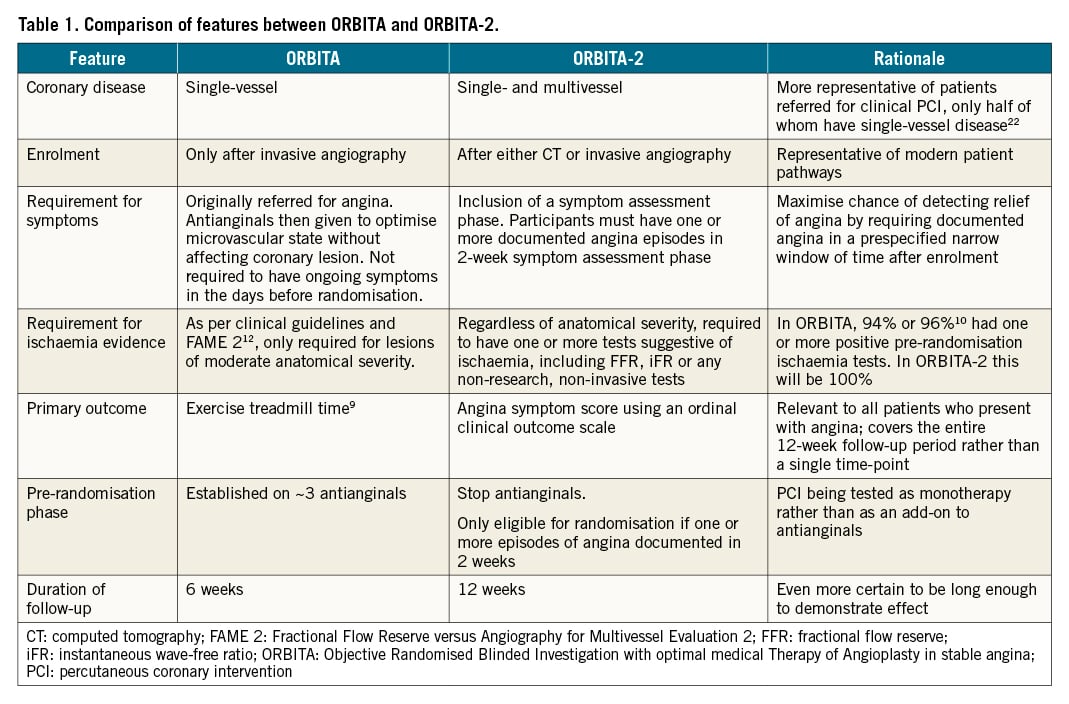
Methods
Study design
ORBITA-2 is a multicentre double-blind randomised placebo-controlled trial to assess the efficacy of PCI for relief of stable angina in single- and multivessel coronary artery disease. The London Central Research Ethics Committee (reference 18/LO/1203) approved the study. The study design is shown in Figure 1.
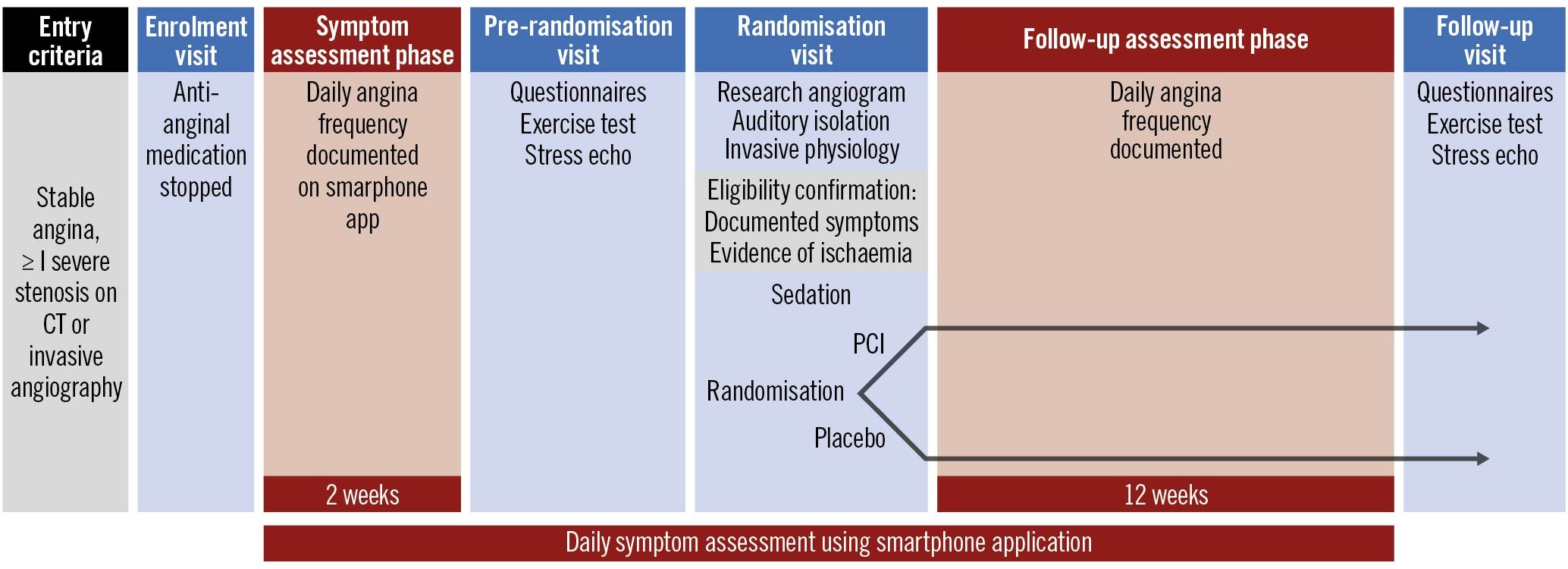
Figure 1. ORBITA-2 study design. PCI: percutaneous coronary intervention; CT: computed tomography
A list of ORBITA-2 investigators is included in Supplementary Appendix 1.
Eligibility criteria
ORBITA-2 will enrol participants who are deemed eligible for PCI by their clinical teams and meet all 3 of the following criteria (Table 2):
1. Angina or angina-equivalent symptoms
2. Anatomical evidence of a severe coronary stenosis in at least 1 vessel on invasive diagnostic angiography or computerised tomography coronary angiography (CTCA)
3. Evidence of ischaemia
For randomisation, the criteria are stricter and the following must also be met:
4. At least 1 episode of angina reported during the 2-week pre-randomisation period
5. Invasive diagnostic coronary angiogram indicating at least one ≥70% stenosis

The exclusion criteria are listed in Supplementary Appendix 2.
By design, some participants will be eligible for enrolment, but will not meet the criteria for randomisation at the time of the research angiogram. For example, a participant may have a severe stenosis on a positive CTCA but have non-flow-limiting disease on invasive physiology at the research angiogram, as judged by the interventionist, or more severe disease necessitating coronary artery bypass graft surgery. As another example, a participant may have an invasive coronary angiogram showing severe disease but have no symptoms during the 2-week symptom assessment phase using the daily smartphone application.
Primary outcome
The primary outcome is an angina symptom score measured daily. This is an ordinal clinical outcome scale designed for assessing health status in angina ranging from 0 to 79, as shown in Supplementary Table 1. The numbers represent a relative ranking without any assumption related to category spacing. It comprises the number of episodes of angina reported daily by the patient on a smartphone symptom application, units of antianginal medications, and high-level category overrides for unblinding due to intolerable angina, acute coronary syndrome and death. Supplementary Table 2 shows the total daily dose of common antianginal medications considered to be one unit.
Participants will be asked once a week to record on the smartphone symptom application whether they had angina during the 2 activities that were described by the participant at enrolment as activities that provoke angina. This is intended to minimise the risk that participants will not exert themselves sufficiently to induce angina and therefore mask their true angina health status.
We surveyed 38 consultant cardiologists and 8 patients for their views on the primary outcome. There was consensus in using an ordinal outcome scale, the ranking of relative worsening of health status in the sequence shown in Supplementary Table 1, and the counting of units of antianginal medications shown in Supplementary Table 2.
Secondary outcomes
- Exercise treadmill time
- Angina severity as assessed by CCS Class
- Angina frequency measured by the symptom application
- Physical limitation, angina stability, quality of life, angina frequency and freedom from angina as assessed with the Seattle Angina Questionnaire (SAQ)
- Quality of life as assessed with the EuroQOL (EQ-5D-5L) questionnaire
- Quality of life as assessed with the MacNew questionnaire
- Breathlessness as assessed with the MRC (Medical Research Council) dyspnoea scale
- Dobutamine stress echocardiography score
- Need for antianginal medication introduction and uptitration
- Unblinding due to intolerable angina
- Admission for acute coronary syndrome (ACS)
- Death
Enrolment
At enrolment, written informed consent will be obtained. Eligibility will be checked. Symptoms will be assessed by CCS class and MRC dyspnoea score. Participants will complete questionnaires including the SAQ, the EQ-5D-5L, the MacNew heart disease health-related quality of life questionnaire and the Rose Angina Questionnaire. They will be taught how to use a smartphone symptom application for recording their symptoms. Patient involvement in the design of the symptom application is described in Supplementary Appendix 3. Participants who do not have a smartphone will be provided with a device and taught how to use it.
The application will notify the research team when participants have failed to report their symptoms. If 3 or more days are missed, participants will be prompted by research staff to enter their symptoms.
Pre-randomisation evaluation
Before randomisation, participants will document symptoms for 2 weeks off antianginals. If they are asymptomatic during this period, they will exit the trial.
Participants will then attend the pre-randomisation visit, where they will have an exercise treadmill test (Supplementary Appendix 4), stress echocardiography (Supplementary Appendix 5), and symptom and quality of life assessment.
Medications
All medication changes will be made by the research team with informed consent from the participant. Decisions will be discussed with primary care practitioners as necessary.
1. Participants not already taking the following medications will be started on:
Dual antiplatelet therapy:
Standard loading doses will be used. Thereafter, aspirin 75 mg once daily with either clopidogrel 75 mg once daily or ticagrelor 90 mg twice daily or prasugrel 5-10 mg once daily, dose adjusted for age and weight, will be administered.
Gastrointestinal (GI) protection:
If at high risk of adverse GI effects (based on previous GI ulceration, age or concomitant medications that increase risk), participants will be started on a proton pump inhibitor, lansoprazole 30mg once daily, in accordance with NICE guidance on gastro-oesophageal reflux disease and dyspepsia in adults (CG184).
Lipid-lowering medication:
Atorvastatin 80 mg once daily will be preferred. If participants are already taking lower dose atorvastatin, simvastatin or pravastatin, this will be changed to atorvastatin 80 mg once daily. If taking rosuvastatin, this will be continued.
2. Other concomitant risk factor modifying medication
Antihypertensives:
Antihypertensives with antianginal properties will be stopped. Participants will be given a blood pressure monitor and asked to perform home readings. Blood pressure control will be monitored by the research team, and if required, antihypertensives will be added. Agents without antianginal properties will be preferred.
3. Antianginal medication
Regular antianginal medications will be stopped on enrolment. All participants will be given glyceryl trinitrate spray to be used when necessary. The need for starting regular antianginals will be determined by participant preference and patient-reported symptoms. An individualised protocol for potential introduction of antianginal medications will be prepared for each participant by the research team. This protocol will be based on the participant's medical history, heart rate, blood pressure and any medication intolerance. The preferred sequence will be as follows: Bisoprolol, nifedipine MR, isosorbide mononitrate MR, nicorandil, ranolazine.
Antianginals started prior to randomisation will be stopped at randomisation and re-introduced according to participant preference and symptoms as described above, by the blinded research team.
Invasive procedure
For the invasive procedure, participants will wear over-the-ear headphones with auditory isolation. Radial or femoral vascular access will be used at the operator’s discretion. Coronary angiography including pressure wire assessment with fractional flow reserve (FFR) and instantaneous wave-free ratio (iFR) will be performed for each ≥50% coronary stenosis, with the pressure wire placed at least 3 vessel diameters beyond the most distal stenosis. Intravenous adenosine will be administered for FFR via an antecubital fossa vein at 140 µg/kg per min. Normalisation will be documented before each measurement. After each measurement, the wire will be checked for drift and, if present, the wire will be renormalised and measurements repeated.
If an operator is unable to pass a pressure wire, no value will be documented, and the images of that case will be published for later verification of anatomic severity.
At this point the operator will select vessels for treatment and the selection will be recorded in the online case report form prior to randomisation. To be eligible for randomisation, there must be evidence of ischaemia on FFR, iFR and/or any non-invasive ischaemia testing performed as part of routine clinical practice prior to enrolment. The non-invasive tests can include cardiac stress perfusion MRI, stress echocardiography or nuclear medicine myocardial perfusion scan depending on local clinical practice. The operators will not have access to the results of the additional stress echocardiography and exercise tests performed as part of the research study. This is an arrangement which preserves the utility of these measures as baseline stratifiers against which effect size can later be regressed1013.
Randomisation and blinding
Randomisation procedures will be conducted at high-volume PCI centres (currently 14 NHS hospital sites across England and Wales). Participants will be sedated, using incremental doses of intravenous benzodiazepines and intravenous opioids, to a deep level of conscious sedation such that they are unresponsive to verbal or tactile stimulus, but airway, ventilation and cardiovascular function are maintained. They will then be randomised 1:1 to PCI or placebo procedure using computer-generated randomisation with block randomisation and block size between 8 and 16 (Randi - opensource clinical trials software). Randomisation will be performed in the catheter laboratory. The allocation will be communicated verbally to the catheter laboratory team. Participants randomised to placebo will be kept sedated in the catheter laboratory for a minimum of 15 minutes post-randomisation.
The participant, and caregivers outside the catheter lab, including ward staff and research staff involved in follow-up assessment and data analysis, will be blinded to treatment allocation. Our protocol will assess for accidental leakage of information to staff and to participants (Supplementary Appendix 6). The blinding index14 will be performed at the time of discharge from the randomised blinded procedure and at follow-up.
Follow-up evaluation
Stress echocardiography, exercise testing and symptom and quality of life assessment will be repeated 12 weeks after randomisation by blinded research staff.
Unblinding and trial end
After the follow-up tests, the participants will be unblinded. Routine medical care will be continued. For avoidance of doubt, research participation will end at the time of unblinding (but ACS and death status will be checked at 12 weeks if early unblinding was performed). This has two implications: first, there will be no merit in “long-term follow-up” beyond this point because unblinded symptom reporting is uninformative and can be misleading; and second, no decision the participant and clinician make at this stage will be considered “crossover” because the participant has exited the trial. Before being randomised the participant was clinically eligible for PCI, had discussed having PCI with their cardiologist, and had agreed to have the procedure. By participating in ORBITA-2, they will have offered researchers the potential to delay their PCI, only for the duration of the trial, and solely to help future participants with angina. It is therefore likely that most participants randomised to placebo will choose to have subsequent PCI.
Data management
Data will be entered onto an electronic case report form (OpenClinica). Data from the smartphone application will be stored on a central server. Data and all appropriate documentation will be stored for a minimum of 10 years after the completion of the study, including the follow-up period.
Data monitoring
All adverse events will be reviewed by the independent Data Safety Monitoring Board (DSMB). The DSMB will also review clinically driven withdrawals and protocolised introduction and uptitration of antianginal medications.
Sample size calculation
The trial is designed to detect a difference between arms in the change in angina symptom score units of one-third of a standard deviation of change in angina symptom units. Using a 2-sample t-test with a 2-sided alpha of 0.05 and 80% power, we calculated that we will need to randomise 284 participants across the active and control arms. Based on the experience of ORBITA we need to allow for a dropout rate of 7% and a crossover rate of 10% from the control arm and 2% in the active arm. This requires 396 enrolled participants. We plan to enrol 400. Crossover refers to a participant being randomised to 1 arm, but then having the treatment specified in the other arm before their participation in the trial ends and they are unblinded at 12-week follow-up. Since all efficacy analysis will be performed on the intention-to-treat basis, such crossover will attenuate the observed effect size.
Recruitment status
Recruitment for ORBITA-2 commenced in November 2018 and was paused in March 2020 due to the COVID-19 pandemic. A protocol amendment was made in May 2020 and recruitment resumed (Supplementary Appendix 7). To date, 217 participants have been enrolled and 130 have been randomised.
Statistical analysis plan
Data will be summarised as quartiles for continuous variables and proportions for categorical ones. Data will be analysed on an intention-to-treat basis. The Bayesian posterior probability of efficacy is the primary evidence summary. The primary outcome of ORBITA-2 is the placebo-controlled efficacy of PCI on the angina symptom score using an ordinal clinical outcome scale for angina. We will use a Bayesian approach with a proportional odds model for the analysis of the primary outcome of angina symptom score adjusted for pre-randomisation angina symptom score. A first-order Markov model will be used to model within-patient correlation in serial measurements. The proportional odds model is efficient in testing the hypothesis while accommodating the statistical distribution and possible floor and ceiling effects. The R rmsb package blrm function will be used for computations15. The statistical model extracts maximum information from the outcome data’s severity and timing of events to maximise power. The rationale for planned stratified analyses is described in Supplementary Appendix 8.
ORBITA was criticised for its statistical methods. A Bayesian approach addresses some of the limitations of p-values. Bayesian posterior probabilities prevent the conclusion of equivalent effects of two treatments on an outcome when the data do not support that interpretation16.
Dissemination plan
As in ORBITA, the primary result in ORBITA-2 will be analysed according to the prespecified analysis plan. Secondary analyses will also be subsequently conducted, testing for dependence of any treatment effect on pre-randomisation indices of ischaemia.
Discussion
ORBITA-2 should provide long-awaited evidence for managing stable angina by assessing the placebo-controlled effect of PCI on angina with no background antianginal therapy. This is important because PCI has associated healthcare costs and small, but non-negligible, risks of short- and long-term complications.
There are 3 principal challenges facing trials of angina treatment. The major challenge is that unblinded angina data are of little or no value. ORBITA and ORBITA-2 resolve this by blinding participants, research, and clinical teams to treatment allocation.
The second challenge is to capture the amount of angina reliably. The historical approach has been a questionnaire which is filled in by the participant at the end of a period, such as a month. This approach has been validated, in the sense that it has shown a correlation coefficient of -0.64 with the gold standard of daily documentation of symptoms17. However, it is limited by recall bias and, more importantly, patients may modify their activity levels resulting in reduced symptom frequency. In ORBITA-2 we take the opportunity of the ubiquitous availability of smartphones to capture this, through gold standard daily documentation. A similar approach was taken in the TERISA (Type 2 Diabetes Evaluation of Ranolazine in Subjects With Chronic Stable Angina) trial, albeit with a non-smartphone electronic device.18. Our daily reporting gives further opportunity to validate the monthly questionnaires and the potential to detect effects with greater temporal precision. Each day the symptom application asks the participant the number of episodes experienced and the intensity of the most severe episode on a visual analogue scale. Additionally, every week the symptom application asks whether the 2 angina-provoking activities that the participant prespecified at enrolment are still causing angina.
The third challenge facing trials of angina treatment was exemplified by Saxon et al, who found that, amongst patients who reported no angina, the clinician documented CCS Class II, III or IV in 20 to 46% of cases19. This may be because staff are influenced by collateral information, such as the anatomical or physiological severity of the lesion and whether it has been treated when grading the CCS. In the DEFER (Deferral of PTCA Versus Performance of PTCA) trial, simply learning that the lesion had an FFR above threshold was enough to reduce the proportion of patients with chest pain from 88% to 54%2021. In FAME 2, the effect of gaining this knowledge was even greater, reducing the proportion of patients with CCS Class II-IV from 67% to 16%1221. ORBITA dealt with this reporting bias by blinding both the participant and the staff member assessing the symptoms at follow-up. ORBITA-2 will do the same.
Limitations
Due to the COVID-19 pandemic, some patients were not able to have exercise testing and stress echocardiography. These assessments were interrupted for a short period of a few months in 2020, when the frequency of enrolment had also decreased, therefore it is unlikely there will be substantial missing data. However, the primary outcome data are remotely collected via the smartphone application, which has therefore been largely unaffected by the impact of the pandemic.
Conclusion
ORBITA-2 will build on the results of ORBITA by assessing the placebo-controlled effect of PCI on angina with no background antianginal therapy in both single- and multivessel disease. Novel features include the use of an ordinal clinical outcome scale for angina and daily symptom reporting using a smartphone application. It will also provide a further opportunity to look for predictors of the placebo-controlled effect of PCI.
Impact on daily practice
ORBITA-2 will provide placebo-controlled data on the efficacy of PCI on symptoms in patients off antianginal medications. An ordinal clinical outcome scale for angina was designed in partnership with patients to be a relevant, informative, and inclusive primary outcome. This outcome incorporates daily documentation of angina episodes on a novel smartphone application. This larger trial, with longer follow-up, will more definitively establish the efficacy of PCI.
Acknowledgements
We acknowledge Nina Bual, Juliet Holmes and Elizabeth Owen for their dedication and support for the trial.
Guest Editor
This paper was guest edited by Franz-Josef Neumann, MD; Department of Cardiology and Angiology II, University Heart Center Freiburg - Bad Krozingen, Bad Krozingen, Germany.
Funding
The study is sponsored by Imperial College London and funded by NIHR Biomedical Research Centre. Philips Volcano (San Diego, CA, USA) provided an in-kind support grant of 400 pressure wires for the study. The sponsor and the funder have no role in the study design: collection, management, analysis and interpretation of the data; writing of the report; or the decision to submit the report for publication.
Conflict of interest statement
R. Al-Lamee receives speaker’s honoraria from Phillips Volcano, Abbott Vascular and Menarini Pharmaceuticals and is on the Advisory board of Janssen Pharmaceuticals. C. Rajkumar is supported by the Medical Research Council. M. Foley is supported by the Medical Research Council and reports speaker’s honoraria from Menarini Pharmaceuticals. J.P. Howard is supported by the Wellcome Trust and received support for attending meetings and/or travel from SCMR Travel Scolarship. A.N. Nowbar is supported by the NIHR Academy. T.R. Keeble has received research support from Abbott Vascular, Medtronic, Terumo and Volcano, reports honoraria from Abbott Vascular and Astra Zeneca, and is Chairman of BCIS OHCA focus group. F.E. Harrell Jr reports consulting fees from Imperial College London. M. Shun-Shin reports honoraria from Pfizer. S. Nijjer reports speaker fees from Philips Volcano, is a committee member of British Cardiovascular Society – unpaid, and President of Cardiology section of the Royal Society of Medicine - unpaid. S.M. Afzal Sohaib is Communications Committee Member of the European Society of Cardiology. H. Selgiman reports fees from Amgen and research funding paid to Imperial College. R. Petraco reports grants from Amgen and consulting fees from Philips Volcano and Amgen. S. Sen reports honoraria from Medtronic, Pfizer, Philips and Edwards Lifesciences. The Guest Editor reports lecture fees paid to his institution from Amgen, Bayer Healthcare, Biotronik, Boehringer Ingelheim, Boston Scientific, Daiichi Sankyo, Edwards Lifesciences, Ferrer, Pfizer, and Novartis, consultancy fees paid to his institution from Boehringer Ingelheim, and grant support from Bayer Healthcare, Boston Scientific, Biotronik, Edwards Lifesciences, GlaxoSmithKline, Medtronic, and Pfizer. The other authors have no conflicts of interest to declare.
Supplementary data
To read the full content of this article, please download the PDF.
