Abstract
Background: More effective and progressively safer generations of drug-elut-ing stents (DES) have replaced bare metal stents (BMS) in rou-tine clinical practice. However, patients considered to be at high bleeding risk (HBR) have traditionally been underrepresented in pivotal DES trials.
Aims: The aim of this study was to model the safety and effectiveness of drug-coated stents (DCS) versus BMS in HBR patients according to the Academic Research Consortium (ARC) criteria.
Methods: Participants from the LEADERS FREE (LF) and LEADERS FREE II (LFII) studies were pooled into one data set. Participants were treated with 30 days of DAPT. The primary safety (composite of cardiac death, myocardial infarction, or stent thrombosis) and effectiveness (target lesion revascularisation) endpoints were compared between DCS and BMS in the subgroup of patients satisfying the ARC-HBR definition using propensity-score modelling.
Results: From the 3,635 participants included in the combined LF and LFII data set, 2,898 (79.7%) satisfied the ARC-HBR criteria (DCS: 1,923; BMS: 975). The primary safety endpoint occurred in 184 (9.8%) and in 132 (13.8%) participants in the DCS and BMS groups, respectively (adjusted HR 0.72, 95% CI: 0.57-0.91; p=0.006). The risk of the primary effectiveness endpoint was also significantly lower with DCS (6.2%) versus BMS (8.8%) (adjusted HR 0.70, 95% CI: 0.52-0.94; p=0.016). The safety and effectiveness of DCS versus BMS were consistent according to ARC-HBR status (p for interaction=0.206 and 0.260, respectively).
Conclusions: DCS are safer and more effective than BMS in an ARC-defined HBR population.
Introduction
More effective and progressively safer generations of drug-eluting stents (DES) have replaced bare metal stents (BMS) in routine clinical practice1. However, patients considered to be at high bleeding risk (HBR) have traditionally been underrepresented in pivotal DES trials. Conceptual concerns about slower endothelialisation with DES mandating longer dual antiplatelet therapy (DAPT) duration have led to the continuous use of BMS in routine clinical practice for HBR patients2. In recent years, the safety and efficacy of contemporary DES designs have been explored in randomised controlled trials focusing exclusively on HBR patients3,4,5,6,7. While the use of identical eligibility criteria across some of these trials promotes informative comparisons, heterogeneous characterisation of the HBR population in the literature complicates the incorporation of study findings into guideline recommendations and clinical practice. This subset of patients carries a high risk of mortality and needs to be well characterised8. Recognising the importance of developing best practice evidence in this high-risk group, a standardised, consensus-based definition was developed by the Academic Research Consortium (ARC) to characterise more consistently the HBR population in future studies9. The ARC-HBR definition is likely to have a strong impact on the designs of future studies but, until now, device safety and comparative effectiveness have never been reported in an ARC-HBR-defined population treated with a one-month DAPT course, and the ability of the ARC-HBR definition to discriminate a true HBR population has only been studied using registry data10,11,12.
Leveraging patient-level data from the LEADERS FREE (LF) and LFII studies, the objectives of this analysis are: 1) to model the safety and effectiveness of drug-coated stents (DCS) versus BMS in a true ARC-HBR population, and 2) to examine the degree to which the ARC-HBR criteria accurately identify a higher bleeding risk subset.
Methods
LF was a double-blind, randomised controlled trial conducted in 68 sites in 20 countries from December 2012 to May 2014. Its methods and results have been published previously3. Briefly, 2,466 participants with an indication for percutaneous coronary intervention (PCI), but considered not to be candidates for prolonged DAPT due to at least one of 13 pre-specified criteria perceived to identify increased bleeding risk (Supplementary Table 1), were randomised to the BioFreedom™ polymer-free abluminal biolimus A9-coated DCS (Biosensors International, Singapore), or to the Gazelle™ BMS (Biosensors). The BioFreedom DCS is a stainless steel BMS platform with a selectively micro-structured abluminal surface allowing the drug (biolimus A9) to adhere to the device without a binder or a polymer, distinct from DES. The protocol mandated DAPT discontinuation 30 days after the index procedure. LFII was a prospective, multicentre, single-arm, open-label pivotal study conducted in 50 centres in the USA, Canada, and Europe5. Eligibility criteria, case report forms, adjudication rules, endpoint definitions, and clinical event committees were identical to the LF trial. All enrolled patients were treated with a BioFreedom DCS, followed by protocol-mandated DAPT for 30 days. Both studies were approved by local institutional review boards, and informed consent was obtained in all patients.
The safety and effectiveness of DCS versus BMS were compared in participants meeting a modified ARC-HBR definition in the pooled LF and LFII study populations. The original ARC-HBR criteria are presented in Supplementary Table 2. Since the LF and LFII studies were designed and conducted before the ARC-HBR criteria were developed, data vocabulary was not identical, and approximations were required. The modified ARC-HBR criteria used for this analysis are presented in Table 1. Also, four ARC-HBR criteria were not captured in the study case report form of the LF and LFII studies: recent major surgery or major trauma within 30 days before PCI, previous traumatic intracranial haemorrhage within the past 12 months, presence of a brain arteriovenous malformation, and chronic bleeding diathesis. Patients meeting at least one major or two minor criteria are considered to satisfy the ARC-HBR definition, developed with the purpose of identifying patients with a ≥4% risk of Bleeding Academic Research Consortium (BARC) bleeding 3 or 5, or a ≥1% risk of intracranial haemorrhage at one year.
The primary safety endpoint is the composite of cardiac death, myocardial infarction (MI), or definite/probable stent thrombosis (ST). The primary effectiveness endpoint is clinically driven target lesion revascularisation (TLR). Both the primary safety and effectiveness endpoints were the same as in the LF trial. All-cause mortality, the individual components of the primary safety endpoint, and the composite of definite/probable ST or MI were examined as exploratory ischaemic endpoints. MI was defined according to the third universal definition of MI, and ST was defined according to the ARC criteria13. TLR was defined as revascularisation for an operator-defined >50% restenosis in the treated lesion in the presence of symptoms or of objective ischaemia, or for a core laboratory-defined restenosis >70% in the absence of symptoms or documented ischaemia. The primary bleeding endpoint for this analysis was type 3-5 bleeding according to the BARC criteria14. Secondary bleeding endpoints were any BARC bleeding, and intracranial bleeding. All endpoints were independently adjudicated. TLR and stent thromboses were reviewed by an independent core laboratory (CERC, Massy, France). All endpoints were reported at one year.
STATISTICAL ANALYSIS
Means and standard deviations are reported for continuous variables and counts and percentages are reported for categorical variables. Baseline characteristics between the DCS and BMS groups were compared using the Wilcoxon rank-sum test for continuous variables, and the chi-square test or Fisher’s exact test for categorical variables. Endpoints were analysed using propensity score in a Cox proportional analysis model to quantify the independent effects of stent type (Supplementary Appendix 1). Hazard ratios (HRs) and 95% confidence intervals (CIs) were reported. The consistency of the results was also evaluated in a sensitivity analysis among ARC-HBR participants of the LF randomised trial only (excluding the LFII participants). Kaplan-Meier analysis and log-rank test were performed to compare the primary safety endpoint between DCS and BMS. The propensity score obtained by inverse probability of treatment weighting was applied to adjust the clinical endpoints of the Kaplan-Meier estimation. The interaction between stent type and ARC-HBR status was evaluated for all the endpoints.
To analyse the overall bleeding rates in the ARC-HBR versus the non-ARC-HBR subgroups, unadjusted rates and HRs (95% CI) for BARC 3-5 bleeding events were reported. A Cochran-Armitage test was used to calculate the p-value for a trend in risk of BARC 3-5 bleeding events according to the number of ARC-HBR criteria. A p-value <0.05 was considered statistically significant. Statistical analyses were performed by Biosensors using SAS 9.4 (SAS Institute, Cary, NC, USA).
Results
PATIENTS
From the 3,635 participants included in the combined LF and LFII populations, 2,898 (79.7%) satisfied at least one major or two minor ARC-HBR criteria and constituted the pooled ARC-HBR cohort (LF: n=1,940; LFII: n=958) (Figure 1). Among the 737 non-ARC-HBR criteria patients, 571 satisfied one minor criterion only, and 166 did not meet any of the ARC-HBR criteria. Mean number of major ARC-HBR criteria was 0.94±0.72 per patient among those meeting the ARC-HBR definition. The most frequent major criteria were long-term use of oral anticoagulants (44.5 %), and haemoglobin <11 g/dL (19.8%) (Figure 2). One-year follow-up was completed in 1,895/1,923 patients (98.5%) with DCS and in 965/975 patients (98.9%) with BMS. Baseline and procedural characteristics according to stent type are presented in Supplementary Table 3.

Figure 1. Study flow chart. ARC-HBR: Academic Research Consortium High Bleeding Risk; BMS: bare metal stent; DAPT: dual antiplatelet therapy; DCS: drug-coated stent; eGFR: estimated glomerular filtration rate; LF: LEADERS FREE; NSAIDs: non-steroidal anti-inflammatory drugs; PCI: percutaneous coronary intervention
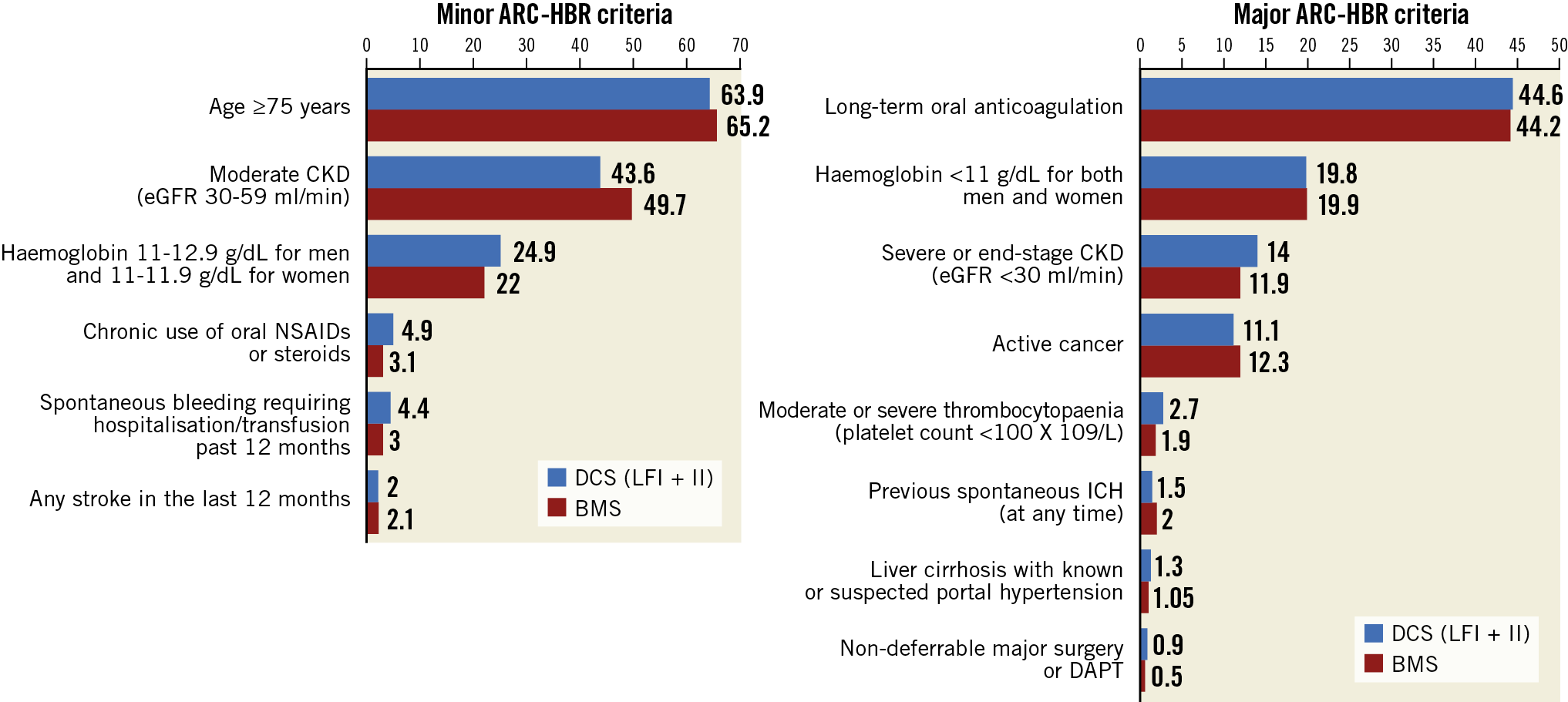
Figure 2. Distribution of ARC-HBR criteria in the pooled LEADERS FREE and LEADERS FREE II studies. ARC-HBR: Academic Research Consortium High Bleeding Risk; BMS: bare metal stent; CKD: chronic kidney disease; DAPT: dual antiplatelet therapy; DCS: drug-coated stent; eGFR: estimated glomerular filtration rate; ICH: intracranial haemorrhage; LF: LEADERS FREE; NSAIDs: non-steroidal anti-inflammatory drugs
SAFETY AND EFFECTIVENESS OF DCS VERSUS BMS IN ARC-HBR PATIENTS
In the ARC-HBR subgroup of the combined LF and LFII cohort, the primary safety endpoint occurred in 184 (9.8%) participants treated with a DCS, and in 132 (13.8%) participants treated with a BMS (adjusted HR 0.72, 95% CI: 0.57-0.91; p=0.006) (Table 2). The treatment effect of DCS on the primary safety endpoint was consistent between the ARC-HBR and non-ARC-HBR subgroups (p for interaction = 0.206). Adjusted Kaplan-Meier analysis of the freedom from the primary safety endpoint according to stent type is presented in Figure 3. The rate of the primary effectiveness endpoint of clinically driven TLR was also significantly lower with DCS (6.2%) versus BMS (8.8%) (adjusted HR 0.70, 95% CI: 0.52-0.94; p=0.016) (Table 2). Only type 1 MIs were significantly reduced with DCS (1.6%) versus BMS (3.8%) (adjusted HR 0.41, 95% CI: 0.25-0.67; p<0.001). The results were consistent in the ARC-HBR subgroup of the LF randomised trial only (Supplementary Appendix 2, Supplementary Table 4, Supplementary Table 5).
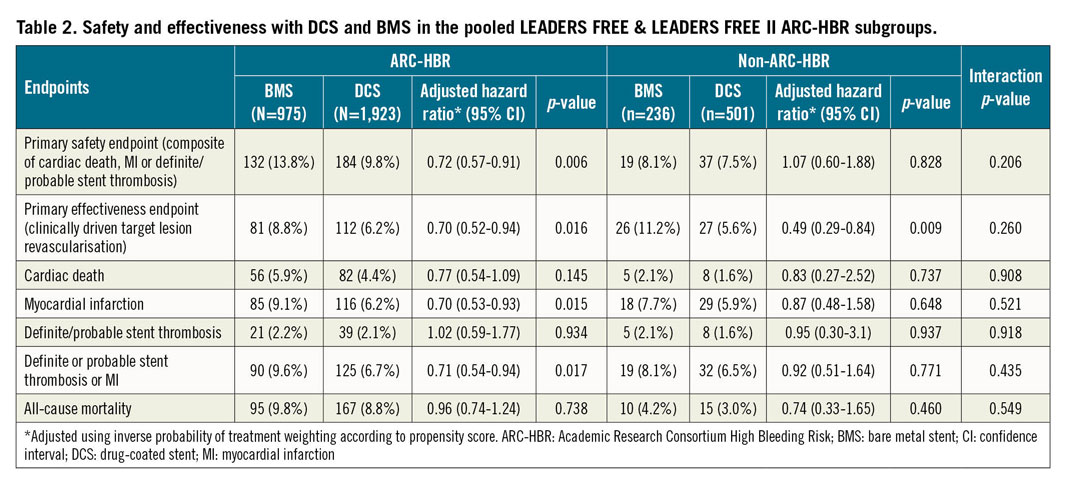
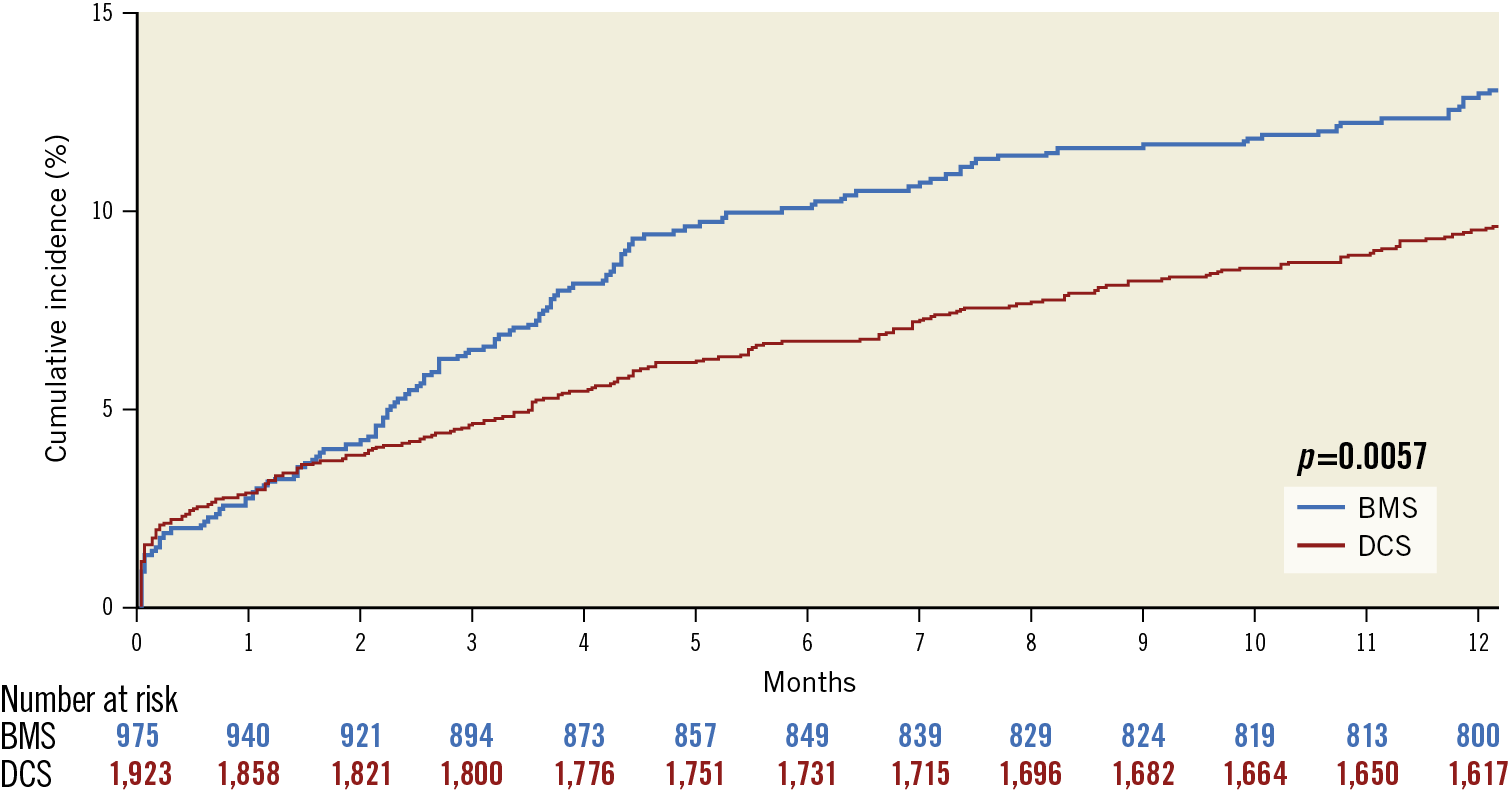
Figure 3. Adjusted Kaplan-Meier analysis of the primary safety endpoint according to stent type for the ARC-HBR subset of the combined LEADERS FREE and LEADERS FREE II studies. BMS: bare metal stent; DCS: drug-coated stent
BLEEDING RISK ACCORDING TO ARC-HBR STATUS
In the pooled LF and LFII cohort, patients meeting the ARC-HBR definition were generally older, more likely to be female, and had more comorbidities compared with those who did not meet the ARC-HBR definition (Supplementary Table 6). A BARC 3-5 bleeding event occurred in 230 (8.2%) ARC-HBR participants, and in 25 (3.5%) non-ARC-HBR participants at one year (HR 2.43, 95% CI: 1.61-3.67; p<0.0001). A BARC 3-5 intracranial bleeding event occurred in 20 (0.7%) ARC-HBR participants, and in 0 (0.0%) non-ARC-HBR participants at one year (HR and p-value not applicable). Kaplan-Meier analysis of BARC 3-5 bleeding events according to ARC-HBR status is presented in Figure 4. Rates of BARC 3-5 bleeding increased significantly across additive ARC-HBR bleeding risk categories, from none (1.9%) to >2 (16.0%) HBR characteristics (p<0.001) (Figure 5). Rates of ischaemic events at one year according to ARC-HBR status are presented in Supplementary Table 7.
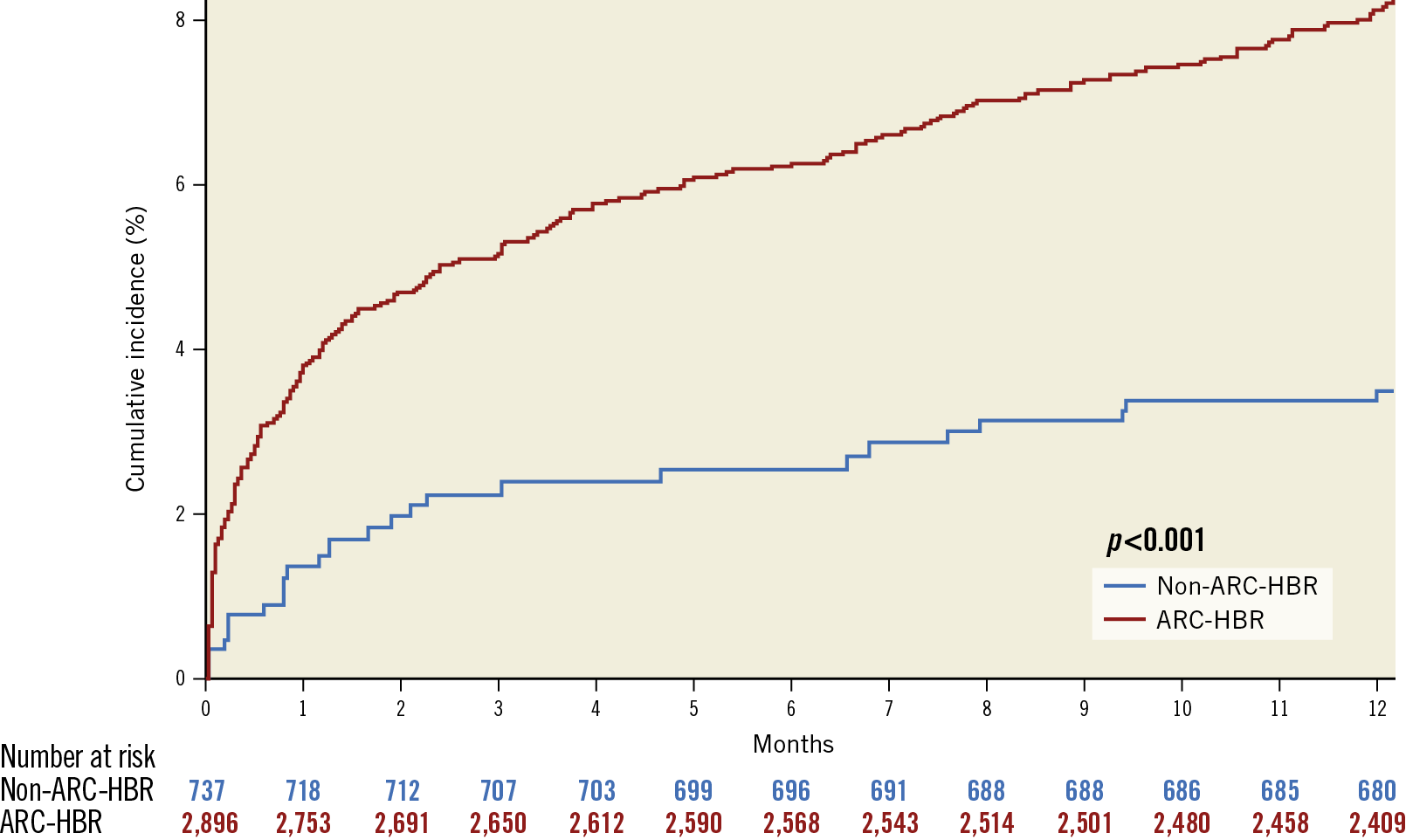
Figure 4. Adjusted Kaplan-Meier analysis of BARC 3-5 bleeding according to ARC-HBR status in the combined LEADERS FREE and LEADERS FREE II studies. ARC-HBR: Academic Research Consortium High Bleeding Risk
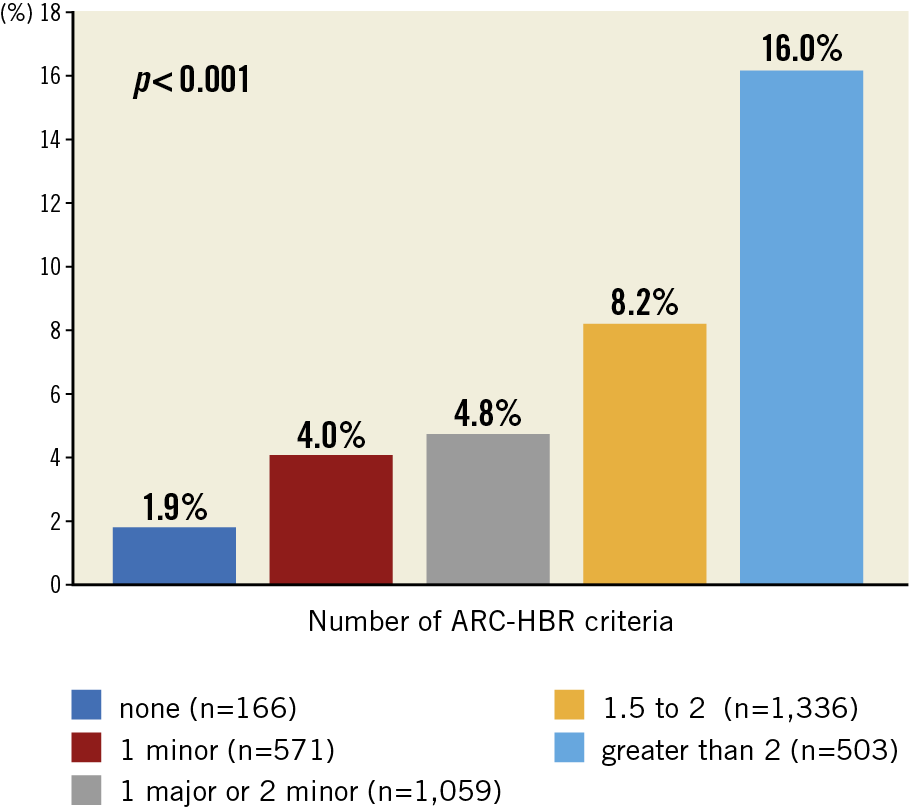
Figure 5. BARC 3-5 bleeding events at one year according to ARC-HBR categories. ARC-HBR: Academic Research Consortium High Bleeding Risk
Discussion
In this study, we present the first report modelling device-related safety and effectiveness after PCI with 30-day DAPT in an ARC-HBR cohort. Our model suggests that DCS are safer and more effective than BMS in the ARC-HBR subgroup. Our results have important implications because, as a pivotal study, LFII positioned the BioFreedom DCS as a predicate for evidence supporting regulatory decisions on benefit/risk with PCI specifically in an HBR population. Multiple other stent platforms are likely to be submitted for similar registration decisions. The similarities and differences between inclusion criteria and outcome results, such as those tested in our model of ARC-HBR cohorts within the LF and LFII studies, will probably be an important dimension of such regulatory decisions as international guidelines are increasingly incorporating ARC-HBR definitions (or regional variations thereof), to guide clinical practice. In routine clinical practice, selecting the optimal device for patients deemed to be at HBR and requiring a short course of DAPT remains challenging in the absence of trials randomising participants based on a standardised HBR phenotype. This population has been the focus of recently published randomised trials aiming to identify stent platforms that would optimally balance safety and efficacy when short DAPT durations are required3,4,5. The ONYX ONE trial reported the non-inferiority, but not the superiority, of the Resolute Onyx™ DES (Medtronic, Minneapolis, MN, USA) in terms of the composite of cardiac death, MI, or ST compared with the BioFreedom DCS in a pre-ARC-HBR population4. Both devices differ in terms of platform, use of polymer, and -limus analogue variation. Based on our model of true ARC-HBR patients within the pooled LF and LFII data set, it seems likely that the results of ONYX ONE will be informative for practice in ARC-HBR patients.
Previous other well-designed trials used inclusion criteria different from ARC-HBR definitions, which confounds turning comparisons of their findings into recommendations for clinical practice. In the Zotarolimus-eluting Endeavor Sprint Stent in Uncertain DES Candidates (ZEUS) trial, patients were randomised to a zotarolimus-eluting stent versus a BMS7. Half of the participants were included on the basis of increased bleeding risk, and the incidence of the composite of death, MI, or target vessel revascularisation was significantly lower with the zotarolimus-eluting stent in this subgroup. In the Short Duration of Dual antiplatElet Therapy With SyNergy II Stent in Patients Older Than 75 Years Undergoing Percutaneous Coronary Revascularisation (SENIOR) randomised trial, the composite of all-cause mortality, MI, stroke, or ischaemia-driven TLR was less frequent with a bioabsorbable polymer DES compared with BMS in individuals ≥75 years old6. Age alone is only a minor criterion for ARC-HBR patient identification, and outcomes of patients in the SENIOR trial who had other ARC-HBR comorbidities have not been reported. Thus, whether the findings of the ZEUS and SENIOR trials will also apply consistently in an ARC-HBR population remains to be demonstrated. The ongoing XIENCE 90 (NCT03218787), XIENCE 28 Global Study (NCT03355742), EVOLVE Short DAPT (NCT02605447), and MASTER DAPT (NCT03023020) studies will also shortly report data on contemporary DES platforms in HBR study populations determined before the publication of the ARC-HBR definition. As the latter is expected to be consistently adopted in the design of future studies with the intention of further clarifying best practices and device and drug benefit/risk in these complex patients, our confirmation that the superior device safety and effectiveness in a pre-ARC-HBR population remains significant in an ARC-HBR subset is reassuring.
Our data also confirm that the ARC-HBR definition accurately dichotomises a group previously perceived as HBR into two subgroups with significantly higher (the ARC-HBR) and lower (the non-ARC-HBR) severe bleeding risks at one year. PCI patients defined by the ARC-HBR criteria indeed met the ARC-HBR threshold of >4% BARC 3-5 bleeding risk at one year, even when treated with DAPT for only 30 days, and a significant additive effect of ARC-HBR major and minor criteria on one-year bleeding rates was observed. While registry-based validation studies of the ARC-HBR criteria have been published previously, they all included patients treated with a standard DAPT duration (vs 30 days in our study), and none have applied ARC-HBR criteria to a device evaluation so far. Interestingly, patients with only one minor ARC-HBR criterion and who did not qualify for the ARC-HBR definition had a risk of bleeding (4.0%) closer to that of those who qualified with one major or two minor criteria (4.8%), than to that of patients without any minor or major ARC-HBR criterion (1.9%). These results, combined with previous validation studies10,11,12 and with further experience with the use of the ARC-HBR criteria, will probably provide a better evidence base to recalibrate the impact of single minor or major criteria and their combinations regarding bleeding risk.
Limitations
Our study has several important limitations. First, this is a post hoc, retrospective analysis in which treatment assignment between DCS and BMS was not randomised within the ARC-HBR subset, and unmeasured confounding might persist despite propensity score adjustment. Second, four major and three minor ARC-HBR criteria needed to be approximated to fit the available information, and four other major ARC-HBR criteria were not available. The latter four criteria are however relatively infrequent in clinical practice, and their exclusion is unlikely to impact on the take-home message of our analysis. Also, the degree to which escalating bleeding risk may also carry escalated thrombotic risk has not been analysed, although important for decision making.
Conclusions
Using high-quality data and adjudicated endpoints from LF and LFII, our study suggests that DCS are superior to BMS in terms of safety and effectiveness in an ARC-HBR population of patients undergoing PCI followed by 30 days of DAPT, consistent with the findings of the LF and LFII trial populations that used pre-ARC-HBR criteria. Our study also shows that the ARC-HBR definition can adequately identify a subgroup of patients with a higher bleeding risk within a cohort already perceived to be at HBR. The consistent use of ARC-HBR criteria in future studies conducted in the intended use HBR population is desirable to facilitate regulatory review, define drug and device benefit/risk profiles, pool study results, and translate findings into clinical practice.
|
Impact on daily practice Selecting the optimal device for patients deemed to be at HBR and requiring a short course of DAPT remains challenging in the absence of trials randomising participants based on a standardised HBR phenotype. We showed that the ARC-defined HBR criteria discriminate adequately patients considered at higher risk of bleeding, and that DCS should be preferred over BMS in this population in clinical practice. Our results have important implications because, as a pivotal study, LFII previously positioned the BioFreedom DCS as a predicate for evidence supporting regulatory decisions on benefit/risk with PCI specifically in an HBR population, and we present the first report modelling device-related safety and effectiveness in a cohort defined by the standardised ARC-HBR criteria. |
Guest Editor
This paper was guest edited by Franz-Josef Neumann, MD; University Heart Centre Freiburg Bad Krozingen, Division of Cardiology and Angiology II, Bad Krozingen, Germany.
>Funding
This analysis was supported by Biosensors Europe.
Conflict of interest statement
G. Marquis-Gravel declares personal fees from Novartis, Servier, JAMP Pharma, Amgen, Alliance BMS-Pfizer, Canadian Heart Research Center (outside the submitted work); and research funding from Bayer, Duke Clinical Research Institute, Université de Montréal, Montreal Heart Institute Foundation, Fonds de Recherche du Québec - Santé (FRQS), and the Canadian Institutes of Health Research (CIHR). P. Urban reports personal fees from Biosensors and Edwards Lifesciences, outside the submitted work, and other from CERC and MedAlliance. S. Copt, S.S. Slama, and H.P. Stoll are full-time paid employees of Biosensors. D. Capodanno and S.J. Pocock report personal fees from Biosensors (outside the submitted work). J.F. Tanguay reports grants from the Duke Clinical Research Institute during the conduct of the study, grants and other from Abbott Vascular, Bayer, and Biosensors, other from BMS-Pfizer Alliance, and grants and other from Novartis outside the submitted work. R. Mehran reports grants, personal fees and other from Abbott Laboratories, grants and other from Abiomed and Bristol-Myers Squibb, grants and personal fees from Chiesi, grants from Applied Therapeutics, AstraZeneca, Bayer, Beth Israel Deaconess, CERC, Concept Medical, CSL Behring, DSI, Medtronic, Novartis Pharmaceuticals, and OrbusNeich, other from Merck, The Medicines Company, Spectranetics/Philips/Volcano Corp, Watermark Research Partners, Claret Medical, and Elixir Medical, personal fees from Boston Scientific, CardiaWave, Janssen Scientific Affairs, Medscape/WebMD, Roivant Services, Sanofi, Siemens Medical Solutions, Medtelligence (Janssen Scientific Affairs), ACC, and AMA, outside the submitted work, and non-financial support and other from Idorsia Pharmaceuticals Ltd., and Regeneron Pharmaceuticals. M.C. Morice is a shareholder and the CEO of CERC, one of the contract research organisations conducting LF and LFII. M.W. Krucoff reports grants and personal fees from Biosensors (during the conduct of the study), and from Abbott Vascular, Medtronic, OrbusNeich, Terumo, and Cordis/Johnson & Johnson (outside the submitted work). The other authors have no conflicts of interest to declare. The Guest Editor reports lecture fees paid to his institution from Amgen, Bayer Healthcare, Biotronik, Boehringer Ingelheim, Boston Scientific, Daiichi Sankyo, Edwards Lifesciences, Ferrer, Pfizer, and Novartis, consultancy fees paid to his institution from Boehringer Ingelheim, and grant support from Bayer Healthcare, Boston Scientific, Biotronik, Edwards Lifesciences, GlaxoSmithKline, Medtronic, and Pfizer.
Supplementary data
To read the full content of this article, please download the PDF.
