
Abstract
Percutaneous coronary intervention with implantation of drug-eluting stents has become the most commonly performed revascularisation procedure in patients with symptomatic coronary artery disease. Continuous iterations of coronary devices incorporating changes in platform materials, geometry, strut thickness, drug release mechanisms and antiproliferative drugs have progressively reduced the rate of device-related adverse clinical events. Objective performance criteria have been proposed for clinical and angiographic outcomes of drug-eluting stents. The rate of device success has been recognised as an intraprocedural endpoint to evaluate the mechanical ability to complete a procedure with the specific device assigned by protocol in randomised comparative trials. The European Commission and the U.S. Food and Drug Administration both provide guidance documents, including the mechanistic evaluation of coronary stents, which recommend operational definitions of device success. While the majority of clinical trials investigating drug-eluting stents have adopted this endpoint definition, inconsistencies in application limit the reliability of comparisons across different trials reporting device success rates. In addition, it is not uncommon that device success rates are not reported by investigators. A consistent definition of device success is essential to allow scientific comparisons of this technical performance endpoint between devices across different trials. Therefore, we performed a systematic evaluation of definitions and reporting of device success in clinical trials. We propose an extended definition as well as considerations for approaching the determination of the device success rates in future percutaneous coronary intervention trials.
Introduction
Percutaneous coronary intervention (PCI) with implantation of drug-eluting stents (DES) has become the most commonly performed revascularisation procedure in patients with symptomatic coronary artery disease (CAD). Iterative developments of coronary devices that introduced changes in platform materials and geometry, strut thicknesses, drug release mechanisms, and antiproliferative drugs have progressively reduced the rate of clinical adverse events. Indeed, current stent technology is regarded as a mature field and it has been proposed that optimal performance criteria might be used to evaluate clinical and angiographic outcomes of new devices1,2. Most contemporary randomised PCI trials compare a novel DES to a current standard-of-care DES, in terms of a device-oriented or patient-oriented composite primary endpoint at one-year follow-up.
Device success rate is an important metric of acute stent performance in clinical trials. Device success is generally defined as the likelihood of completing the goal of the PCI procedure – to reduce a coronary obstruction to non-obstructive severity – using the specific stent allocated by the trial protocol. In addition to the stent design per se, device success may also be affected by the stent delivery system.
Device success is often reported in conjunction with a procedure success endpoint, which reflects that the lesion treatment result is achieved without doing harm to the patient. Assessment of device success is important in the evaluation of a new stent technology. Regulatory authorities, such as the European Commission and the U.S. Food and Drug Administration (FDA), provide guidance documents for the mechanistic evaluation of coronary stents which recommend an operational definition of device success as an acute device performance endpoint.
Although derivatives of this definition are frequently used in PCI trials, variable implementation and inconsistencies in adjudication limit the capability of meticulous comparisons across different trials reporting on device success rates. In addition, it is not uncommon that device success rates are not reported by investigators. A consistent definition is essential to allow scientific comparisons of this intraprocedural endpoint across different trials reporting on different devices or in different patient populations or coronary anatomy.
In contemporary trials, device failure rates range from <1% to 5%3,4. Whether observed variations in acute device success are in fact device design related or trial ascertainment dependent is uncertain and provides a rationale for the development of more consistent approaches and definitions. In the setting of low one-year clinical outcome event rates across contemporary stent platforms, more technical stent features such as deliverability reflected by device success may play a greater role for operators selecting stents for clinical practice.
We performed a systematic review to evaluate device success rates and definitions in clinical trials with broadly inclusive patient recruitment published in leading cardiology journals, based on critical appraisal of the literature, and summarised case examples of device failure. A summary of the literature search strategy and results is shown in Supplementary Table 1 and Supplementary Figure 1. We propose a standardised extended definition of device success for future PCI trials.
DEFINITION OF DEVICE SUCCESS
In 2008, the European Commission and the U.S. FDA published guidance documents for non-clinical and clinical evaluations of DES to provide recommendations to manufacturers and notified bodies5,6. In the guidance document published by the European Commission, device success is defined as successful delivery and deployment of the device and attainment of <50% diameter stenosis using only the study device (Table 1). Procedure success must meet the angiographic criterion of device success plus additional criteria related to the clinical outcome of the procedure regardless of whether the protocol-assigned device is used. In cases of multiple lesion treatment, all treated lesions must meet the clinical procedure success.

More specifically, procedure success requires the absence of ischaemia-driven adverse events during the hospital stay up to a maximum of the first seven days after the index procedure5. These adverse events include all-cause death, any myocardial infarction (including periprocedural), all coronary revascularisations (target lesion revascularisation, target vessel revascularisation or non-target vessel revascularisation), and coronary device thrombosis. The U.S. FDA Guidance for Industry on Coronary Drug-Eluting Stents does not refer specifically to device success, but rather to scenarios of device malfunction which correspond to device failure. A malfunction is defined when the device does not meet its performance specifications which include all claims made in the labelling for the device. This approach requires consistency throughout the labelling process for coronary stents.
In 2013, the European Society of Cardiology (ESC) was requested by the European Commission to make recommendations for a revision of the European Union medical device advisory document on the evaluation of coronary stents. This work was carried out by a Task Force established by the European Association of Percutaneous Cardiovascular Interventions (EAPCI)1. The document summarises the process required for regulatory and market approval for coronary stents in Europe. It offers the basis for establishing objective performance criteria for clinical and angiographic outcomes when evaluating new devices. A revision of the guidance document has not been published to date, though the recommendations of the Task Force are likely to be taken into account in preparing a new broadly similar type of document for coronary stents known as a Common Specification, which is at draft stage. Furthermore, the Task Force was not asked to propose a standardised methodology for the assessment of device success. In September 2018 the FDA announced a public consultation concerning their guidance document, which is being updated7.
The Academic Research Consortium (ARC) consensus documents for clinical endpoint definitions in coronary stent trials8,9 do not include definitions of device success as it is a mainly technical endpoint.
DEVICE SUCCESS IN ALL-COMERS TRIALS
Device success rate is usually reported in the first-in-human or pilot study results when testing new coronary devices. However, the results from such studies frequently lack generalisability due to the small sample size and the inclusion of highly selected patients with less complex lesions. A comprehensive evaluation of device success can be further substantiated in pivotal trial designs, which typically include a substantial number of patients and are powered to evaluate clinical endpoints. Moreover, trials with an “all-comers” design denoting inclusion of patients across the spectrum of clinical presentation and lesion complexity and more representative of those encountered in real-world practice have been introduced in the evaluation of coronary stents10. Supplementary Table 2 summarises the definitions and success rates of devices used in all-comers trials.
Most PCI trials reporting on device success adopted the definition recommended by the European Commission, but it is noteworthy that device success rates are not reported in the same fashion and are not reported in all studies. The most common variation is the definition of final in-stent residual stenosis, which ranges from <20% to <50%. According to the principal angiographic endpoints recommended by the ESC/EAPCI Task Force on the evaluation of coronary stents1, a post-procedural residual stenosis should be <20% as assessed by coronary angiography. It has been shown that in-stent stenosis ≥20% is associated with an increased risk of target lesion revascularisation11. Since visual estimation of coronary cineangiograms could have high interobserver and intraobserver variability12,13, quantitative coronary angiography (QCA) is a preferred methodology for adjudication of device success.
In the SORT OUT (Scandinavian Organization for Randomized Trials with Clinical Outcome) trials (III, IV, V and VI), the term “device failure” was used instead of device success. The definition of device failure was stated, but the results were not provided14,15,16,17. In the BIOSCIENCE (Ultrathin Strut Biodegradable Polymer Sirolimus-Eluting Stent versus Durable Polymer Everolimus-Eluting Stent for Percutaneous Coronary Revascularization) trial, the first large randomised trial of a thin-strut cobalt-chromium sirolimus-eluting stent with a biodegradable polymer, compared to the XIENCE stent (Abbott Vascular, Santa Clara, CA, USA), the device success rate was also not reported18. In the SORT OUT VII trial, only the rate of device delivery failure was mentioned (Orsiro [Biotronik, Bülach, Switzerland] 1.6% vs Nobori® [Terumo Corp., Tokyo, Japan] 1.7%)19; the definition of device delivery failure was not provided.
Recently, the TARGET (Targeted therapy with a localised abluminal groove, low-dose sirolimus-eluting, biodegradable polymer coronary stent) all-comers trial was published. It compared a low-dose sirolimus-eluting stent, Firehawk® (MicroPort, Shanghai, China) to the XIENCE stent. The device success rate of Firehawk was significantly lower than that of XIENCE (Firehawk 92.4% vs XIENCE 94.8%, p=0.025)20. The device success rate in the XIENCE group in the TARGET trial was numerically lower than that reported in most previous all-comers trials. In the Firehawk group, 2.1% of lesions were treated by a non-assigned stent, 0.7% of lesions were crossover to the XIENCE stent, and 2.5% of lesions were not treated by stent implantation. These differences suggest a difference in the performance of the device during the index procedure. However, possible alternative explanations could be unbalanced differences in any of the following: protocol violation, assigned device not available, change of indication for stent implantation (e.g., patient was referred to surgery), PCI procedure failure (e.g., wire or balloon failed to cross target lesions), or failure to deliver or deploy the assigned device. In fact, the reported unsuccessful study-stent implantation rate of Firehawk was only 0.9%.
Several factors affect the device success rate, including anatomical aspects, lesion characteristics, experience and blinding of operators, properties of both the stent and delivery system (balloon) design as well as the definition and reporting of device success. Theoretically, the influence of these factors would be minimised and balanced in a randomised trial. Thus, the device success rate of coronary devices could be attributed to the trackability, crossability, and pushability of the device. An increasing number of manufacturers are investing in and developing coronary stent/scaffold platforms worldwide. In this context, clinicians and trialists should maintain a high degree of attention to device success rates in PCI trials, as problems may first come to light after more widespread clinical use and investigation in post-marketing studies.
SPECIAL CONSIDERATIONS
LESION-BASED ANALYSIS ISSUES
In contemporary large-scale PCI trials, device success rate is usually reported by a lesion-based analysis, meaning that the numerator and denominator represent the number of lesions, instead of the number of devices. We propose to use the denominator for device success from “per-protocol” analysis, i.e., the denominator should consist of all target lesions where the assigned device is at least attempted once before any other non-protocol therapy. Therefore, target lesions for which PCI was not attempted, no device implantation was attempted (e.g., failure to cross the lesion with guidewires or lesions treated only with balloon angioplasty due to small vessel diameter or restenosis) and non-assigned device implantations without attempt to use assigned device (e.g., assigned device was transiently not available “on the shelf”) would be excluded from the denominator for device success. This per-protocol analysis could ascertain more accurately the technical performance of the device. However, it may be at variance with the conventional intention-to-treat analysis, particularly if the operator changes his intention-to-treat without testing the assigned device (e.g., when considering challenging lesions or for whatever reason). Therefore, detailed explanation of the changes of device selection need to be captured (mandatory). An intention-to-treat analysis for device success could be considered as a sensitivity analysis to assess whether all the intended devices have indeed been implanted. A comparison of analytic methods for device success is provided in Table 2 and Supplementary Figure 2.
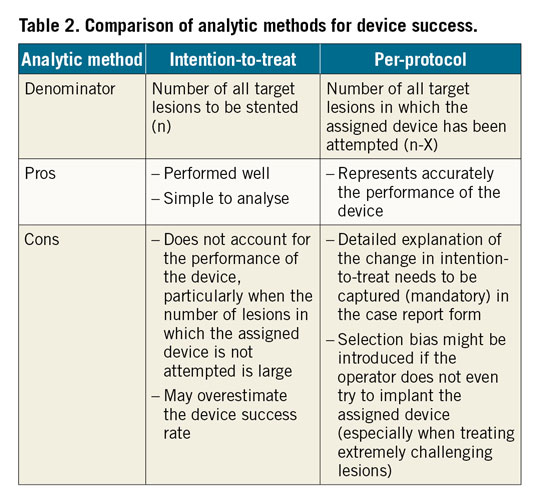
USE OF MULTIPLE DEVICES
Another scenario to be considered for more consistent reporting is the use of multiple devices in one target lesion. For instance, the first assigned device could not cross the target lesion due to inadequate lesion preparation or less deliverability of the device and subsequently the device (stent and its delivery system) was damaged or dislodged. Then, a second assigned stent was eventually implanted after more aggressive balloon predilatation. Normally this scenario would be considered as a device success. However, it might be informative to subclassify device success per protocol according to the number of assigned devices failing (replaced) before final deployment in the lesion. Another approach was seen in the AIDA (Amsterdam Investigator-initiateD Absorb strategy all-comers) trial, where the investigators specified that the device success rate should be counted solely on the basis of the first assigned stent or scaffold21. Therefore, we suggest that the unplanned use of a second (or more) assigned device due to the failure of the first device should be considered as device failure irrespective of the mechanism (related to the device, the lesion or the operator). Health economic reasons (e.g., cost of the second device) could be put forward to rationalise and justify this recommendation. Careful consideration of additional scenarios is required if this approach is taken. For instance, on the basis of lesion-based analysis, when treating a long diffuse lesion or a bifurcation lesion, where implantation of two (or more) assigned devices is planned, the implanted devices should be assessed and reported as one, and all must be successfully implanted to meet the criteria of device success.
MULTIPLE LESIONS IN THE SAME VESSEL
Another scenario to be considered is when multiple target lesions exist in one target vessel, for instance, in the presence of a proximal and a distal lesion in the left anterior descending artery. Assume, for example, that the operator was intending to deliver an assigned device to a distal lesion first and had prepared both lesions for delivery. However, the operator failed to cross the proximal lesion with the assigned device, but then successfully implanted non-assigned devices in both the proximal and the distal lesions. Whether the treatment of the proximal lesion should be counted as device failure and the treatment of the distal lesion should be excluded from the denominator for device success analysis, or whether both lesions would be counted as device failures should have clear definition in the protocol or adjudication rules.
LESION SUCCESS VERSUS DEVICE SUCCESS
Lesion success rate, as opposed to device success rate, has been reported differently in several trials and requires a brief discussion. In the DUTCH PEERS (DUrable polymer-based sTent CHallenge of Promus ElemEnt versus ReSolute integrity) trial, lesion success was defined as the attainment at the target site of a final residual diameter stenosis of less than 50% by any percutaneous method22. In the BIOFLOW V (Ultrathin, bioresorbable polymer sirolimus-eluting stents versus thin, durable polymer everolimus-eluting stents in patients undergoing coronary revascularisation) trial, lesion success was also reported in a similar fashion with final diameter stenosis less than 30%23. In some scenarios, the treating physicians might change their mind during the procedure and decide not to implant the assigned device at the target lesion (e.g., small vessel diameter, in-stent restenosis, unsuccessful angioplasty, coronary slow-flow phenomenon). Thus, reporting lesion success rates provides complementary information on top of device success rates in PCI trials.
Nevertheless, one major limitation of the definition of lesion success is that it is based on the visual angiographic residual severity of the lesion after interventions, without any objective quantitative information such as the assessment of coronary blood flow. Since thresholds of fractional flow reserve (FFR) and instantaneous wave-free ratio (iFR) pressure-derived parameters have been identified, and since the angiography-derived quantitative flow ratio (QFR) might become available for every treated vessel (lesion) by off-line analysis by a core laboratory, additional consideration should be given to an intraprocedural vessel-oriented composite endpoint. Such a definition may require further consensus and is beyond the scope of this document.
RECOMMENDATIONS FOR FUTURE PCI TRIALS
The introduction of coronary stents for the treatment of CAD was accompanied by important developments in clinical research and trial conduct. Numerous clinical trials have been conducted to investigate new stent technologies in a protocolised and, as much as possible, standardised manner. Therefore, an extended definition of device success and a standardised methodology for assessing and reporting this acute performance endpoint in PCI trials are timely. The proposed extended definition is presented in Table 1.
Figure 1 illustrates a practical, mutually exclusive and chronological algorithm for analysing the device success rate. Step 1 establishes the denominator consistent with a “per protocol” analysis, and is the most crucial aspect of device success to decide upon for any given trial design. This denominator definition has a great influence on the device success rate. We suggest preferentially excluding target lesions where no implantation of the assigned device was attempted. The denominator of the device success rate would thus be n-X, representing the number of lesions in which the operators did try to implant the assigned device. Step 2 identifies lesions for which no device (either the assigned or any non-assigned device) was implanted despite single or multiple attempts. Step 3 indicates lesions for which devices were implanted outside the intended location (e.g., device dislodgement with either deployment or crush of the device). Step 4 indicates lesions in which non-assigned devices or second (or more) assigned devices were implanted, because of the unsuccessful delivery or deployment or damage to the first assigned device (e.g., tortuosity/non-crossability of the lesion or defective delivery system/balloon). Lastly, step 5 excludes lesions with final in-stent/scaffold residual stenosis equal to or above 20% by QCA (preferred methodology) or by visual estimation if QCA is not available, along with the recommendation that final data reported for the trial ideally would rely on core laboratory QCA of the final residual stenosis.
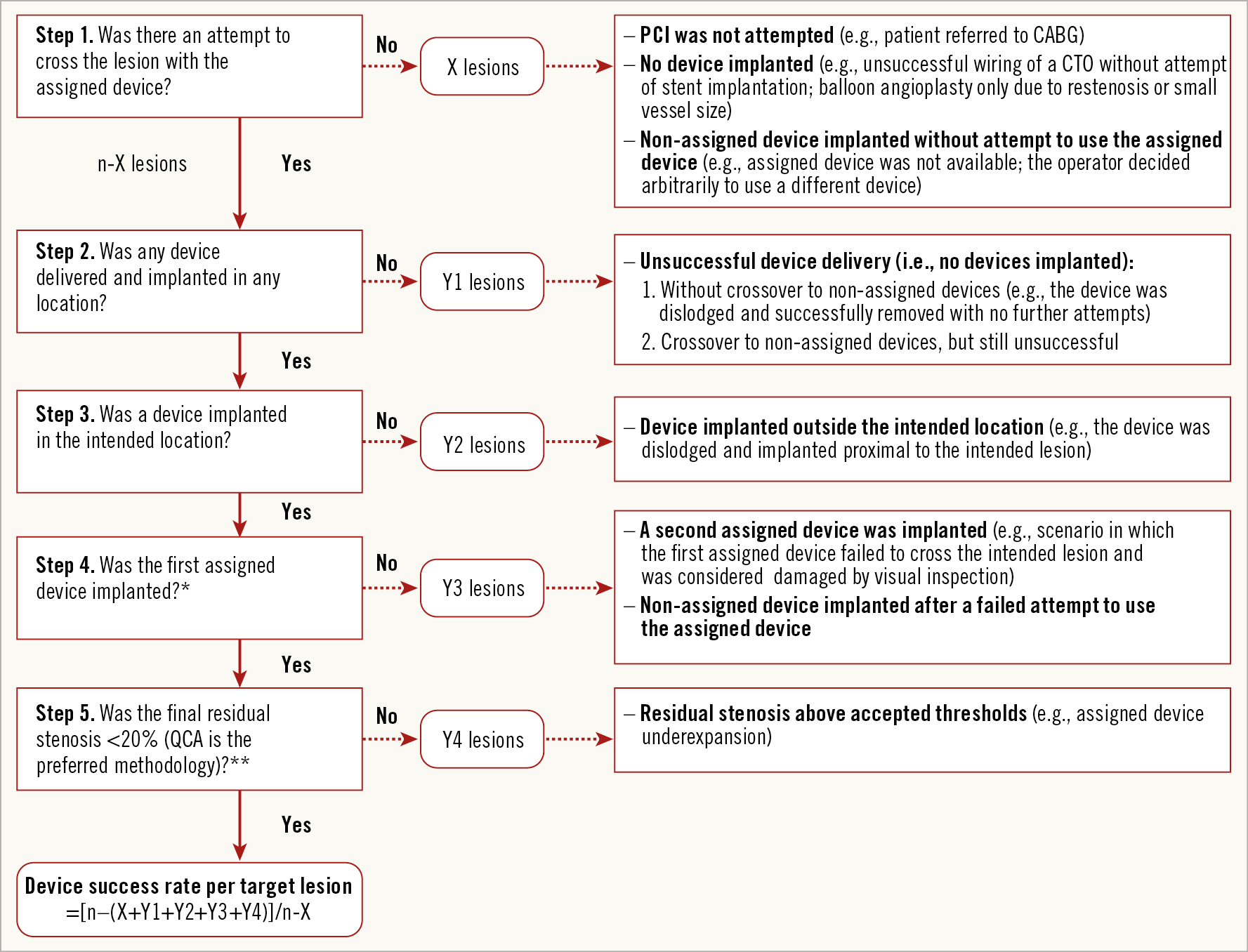
Figure 1. Algorithm for device success analysis. * Please refer to Table 1 if more than one assigned device is planned upfront for one target lesion and Supplementary Figure 3 for details on potential scenarios. ** Additional intravascular image may be useful to confirm the stent deployment, particularly when interpretation of final angiography is limited (e.g., tortuosity or angulation of the vessels, artery overlap, or no-reflow phenomenon) after stent implantation. CABG: coronary arterial bypass grafting; CTO: chronic total occlusion; n: total lesion number; PCI: percutaneous coronary intervention; QCA: quantitative coronary angiography
Figure 2 summarises two examples of device failure and demonstrates the importance of knowing whether the assigned stent was attempted or not, in order to assess the device success rate. Potential scenarios after unsuccessful delivery of a first assigned device to the intended lesion are summarised in Supplementary Figure 3.
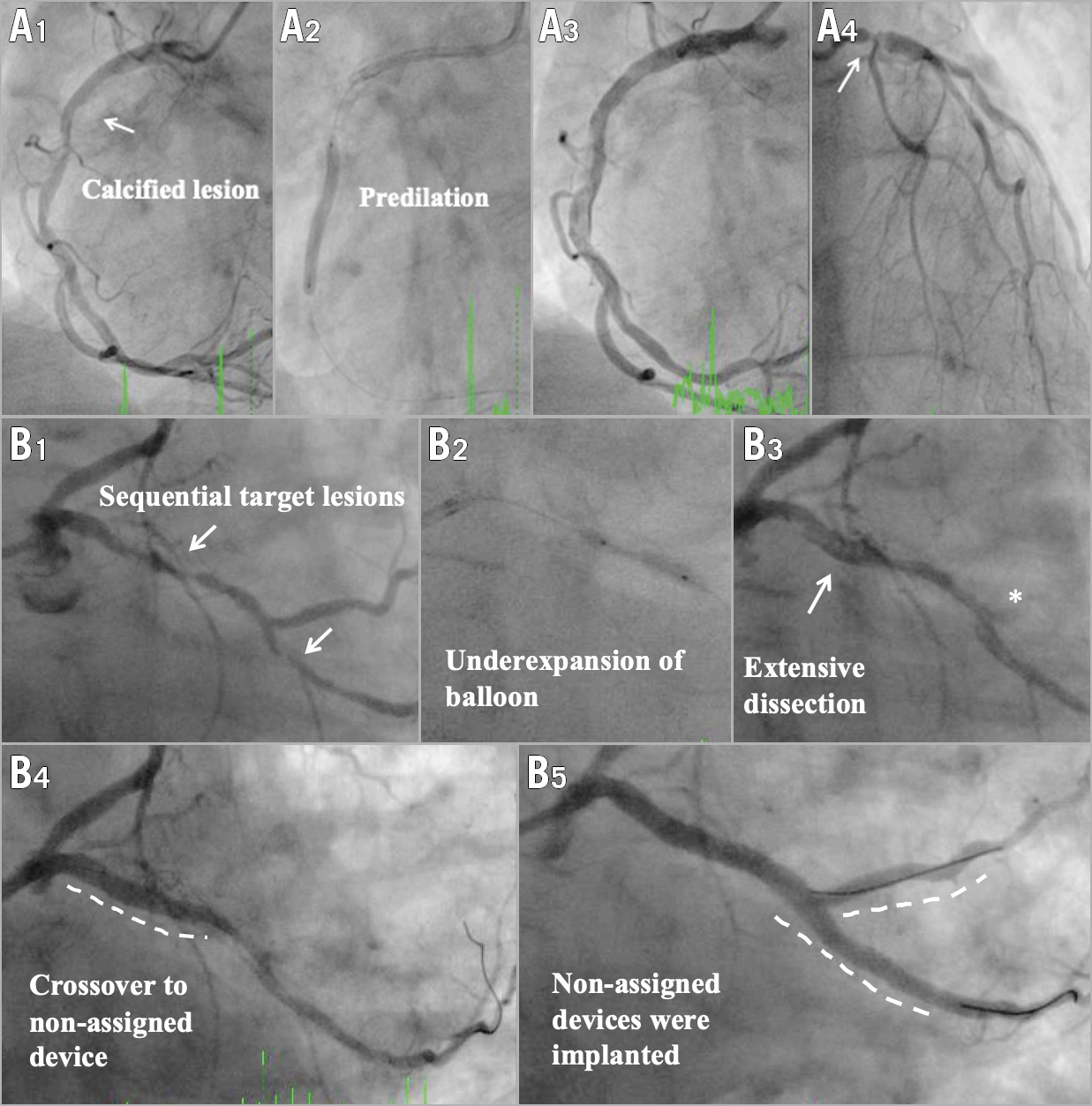
Figure 2. Examples of device failure in the device success analysis. A1. One calcified lesion in right coronary artery (segment 2). A2. Predilation. A3. Assigned device failed to cross the calcified lesion. No device was implanted. A4. Because the assigned device failed to cross the lesion in the right coronary artery, the operator decided to use a non-assigned device for treatment of left main stem (segment 5). Segment 5 should be excluded from the denominator in the analysis of device success rate. Changes in intention-to-treat should be specifically reported in the case report form. B1. Two sequential lesions in left circumflex artery (segments 11 & 13). B2. Predilation. B3. Extensive dissection with compromised blood flow of side branch (*segment 12b). B4. Assigned device failed to cross the proximal lesion, then crossover to a non-assigned device. B5. The operator decided to use non-assigned devices for treatment of the distal lesion and the side branch. Segment 13 should be excluded in the analysis of device success rate. Segment 12b should be reported as a procedural complication with bail-out stenting of assigned device group based on intention-to-treat. Coronary artery segments are defined according to the American Heart Association classification.
Issues of potential operator bias in the assessment of device success must also be considered. In current stent trials investigating two different platforms or iterations, it is not possible to mask the operator who can recognise the commercial products. Behavioural differences based on the experience of the operator cannot be avoided. In the TALENT study24, more crossover to non-assigned stents occurred in the investigational group. Interestingly, this phenomenon was also observed in the TARGET study. It suggests that operators might tend to crossover quickly to the device with which they are familiar when facing difficulties during the PCI procedure, especially in treating patients with multivessel disease. In the TALENT study, the crossover to non-assigned stents was clustered in seven out of 23 centres and was related to the lesion complexity, PCI volume and possibly to the expertise of the operators.
In addition, it is also possible that the assigned device per randomisation is not available “on-shelf” at the time of the procedure, or that the available sizes and lengths of the investigational platform offer fewer options than the comparator, increasing the chance of crossover to the comparator. Alternatively, the new device may be truly less effective in crossing lesions, requiring more frequent crossover. Differentiating between these possibilities is problematic without detailed questioning in the case report form. It is acknowledged that accurate determination of this more granular, consistent and informative approach to device success will challenge existing clinical trial processes to include site work documentation, monitoring visits to ensure accuracy, and a simple, flexible case report form for study use. We propose an example of a case report form that will capture important parameters for the adjudication of device success in PCI trials (Supplementary Table 3).
FUTURE PERSPECTIVES OF DEFINING DEVICE SUCCESS
Stent underexpansion is usually defined according to the diameter stenosis after the procedure, measured by QCA. Coronary angiography only assesses residual stenosis which can be influenced by many factors (e.g., plaque prolapse). The discrepancy in QCA between metallic DES and polymeric bioresorbable scaffolds has been reported25. Intracoronary imaging, such as intravascular ultrasound or optical coherence tomography, provides more accurate ascertainment than angiography in optimising PCI procedures and improves clinical outcome26,27. The use of intracoronary imaging might be a preferred method rather than QCA to assess acute performance of devices for state-of-the-art trials.
Computational fluid dynamic models have shown that, even at the same diameter stenosis, anatomic differences such as stent eccentricity affect local haemodynamics which are related to stent restenosis28. On the other hand, post-PCI FFR has been shown to be a predictor of long-term outcome29,30. Suboptimal stent deployment is known to be associated with a trans-stent FFR gradient after PCI31. Local haemodynamics and functional assessment of stented vessels might become alternative approaches to evaluate the acute performance of devices. Nevertheless, the systematic use of intravascular imaging or FFR after PCI needs to be balanced with risks and costs. In this context, the use of angiography-derived functional assessment such as QFR that does not require the use of additional catheters32,33,34 will probably become widely available in catheterisation laboratories and play an increasing role in the assessment of device success, including the independent assessment by core laboratories. The additional value versus the cost of such data collection enhancements may vary across different study design applications.
Conclusions
Between devices with similar long-term clinical outcomes, device success rates may convey important information for operators choosing devices in clinical practice. Consistent approaches and definitions for device success may greatly enhance the value of such data. This document proposes a feasible approach summarised in a simple algorithm which, if embraced by international cardiovascular societies and clinical research organisations, will allow meaningful comparisons among future studies and advance regulatory science for informative device evaluation.
Appendix. Authors’ affiliations
1. Thoraxcenter, Erasmus University Medical Center, Rotterdam, the Netherlands; 2. Division of Cardiology, Department of Internal Medicine, Taipei Veterans General Hospital, Institute of Clinical Medicine, National Yang Ming University, Taipei, Taiwan; 3. Academic Medical Center, University of Amsterdam, -Amsterdam, the Netherlands; 4. Cardialysis B.V., Rotterdam, the Netherlands; 5. Department of Cardiovascular Diseases, Deutsches Herz-zentrum München, Technische Universität München, and DZHK (German Centre for Cardiovascular Research), partner site Munich Heart Alliance, Munich, Germany; 6. Division of Cardiology, C.A.S.T., P.O. “G. Rodolico”, Azienda Ospedaliero-Universitaria “Policlinico-Vittorio Emanuele”, University of Catania, Catania, Italy; 7. Swiss Cardiovascular Center Bern, Bern University Hospital, Bern, Switzerland; 8. Baim Institute for Clinical Research, Boston, MA, USA; 9. Duke Clinical Research Institute, Durham, NC, USA; 10. New York-Presbyterian Hospital/Columbia University Medical Center, New York, and Clinical Trials Center, Cardiovascular Research Foundation, New York, NY, USA; 11. Barts Heart Centre, London, and Queen Mary University of London, London, United Kingdom; 12. Section for Cardiology, Yale University School of Medicine, New Haven, CT, USA; 13. The Zena and Michael A. Wiener Cardiovascular Institute, The Icahn School of Medicine at Mount Sinai, New York, NY, USA; 14. School of Medicine, Cardiff University, University Hospital of Wales, Cardiff, United Kingdom; 15. Department of Cardiology, Barts Heart Centre, Barts Health NHS Trust, London, United Kingdom; 16. NHLI, Imperial College London, London, United Kingdom.
Guest Editor
This paper was guest edited by Alec Vahanian, MD, PhD; Department of Cardiology, Hôpital Bichat-Claude Bernard, and University Paris VII, Paris, France.
Conflict of interest statement
Y. Onuma was an advisory board member of Abbott Vascular. R.A. Byrne reports personal fees from B. Braun Melsungen AG, and Biotronik, grants and personal fees from Boston Scientific, grants from CeloNova Biosciences and Micell Technologies, outside the submitted work. G.W. Stone reports personal fees from Terumo, Amaranth, Shockwave, VALFIX, TherOx, Reva, Vascular Dynamics, Robocath, HeartFlow, Gore, Ablative Solutions, Matrizyme, Miracor, Neovasc, V-Wave, Abiomed, Claret, BackBeat, Sirtex, and MAIA Pharmaceuticals, personal fees and other from Ancora, Qool Therapeutics, and SpectraWAVE, other from Cagent, Applied Therapeutics, Biostar family of funds, and MedFocus family of funds, outside the submitted work; Columbia University, his employer, receives royalties from Abbott for sale of the MitraClip. A. Baumbach reports grants and personal fees from Abbott, personal fees from AstraZeneca, Sinomed, MicroPort, and KSH, outside the submitted work. P.W. Serruys reports personal fees from Abbott Vascular, Boston Scientific, Biosensors, Medtronic, Philips/Volcano, Sino Medical Sciences Technology, SMT and Xeltis, outside the submitted work. The other authors have no conflicts of interest to declare. The Guest Editor is a consultant for Edwards Lifesciences.
