Introduction
Percutaneous left ventricular assist devices (pLVAD) are indicated to provide short-term mechanical circulatory support in patients with cardiogenic shock, acute myocardial infarction (AMI) with cardiogenic shock, and for high-risk percutaneous coronary intervention (HRPCI)1. The Impella® device (Abiomed Inc., Danvers, MA, USA) is a non-pulsatile microaxial flow pump that continuously propels blood from the left ventricle (LV) to the ascending aorta. The Impella system is placed retrogradely across the aortic valve under fluoroscopic guidance, with its inflow in the LV and outflow in the ascending aorta. The Impella platform consists of several different models that vary in calibre, insertion technique, and maximum haemodynamic support capabilities. There are limited published post-approval surveillance data on the most commonly reported complications and failure modes associated with the Impella devices. We analysed the US Food and Drug Administration (FDA) Manufacturer and User Facility Device Experience (MAUDE) database to report these endpoints.
Methods
The MAUDE database is a searchable online repository created by the FDA to capture major adverse events involving medical devices2. MAUDE reporting can be mandatory (for manufacturers and device user facilities) or voluntary (for healthcare professionals, patients, and consumers). Established in the 1990s, the database is updated monthly, and each medical device report (MDR) contains information on the device, event date, whether the device was returned to the manufacturer, date returned, and description of the event by the user and manufacturer. Based on their severity, events are classified into four categories: death, injury, malfunction, or other. The database was last accessed on 31 August 2018. Two independent reviewers quarried the database from 1 August 2008 to 31 August 2018 for Impella devices, yielding 448 medical device reports. After excluding the Impella RP and incomplete reports, 407 reports were included in the final analysis. Percentages represent the proportion of total submitted MAUDE reports.
Results
The most commonly reported Impella type in our analysis was the Impella CP (Figure 1). Of the 407 MDRs, 131 lacked information regarding clinical indication. Impella devices were most commonly placed for HRPCI (Supplementary Table 1).
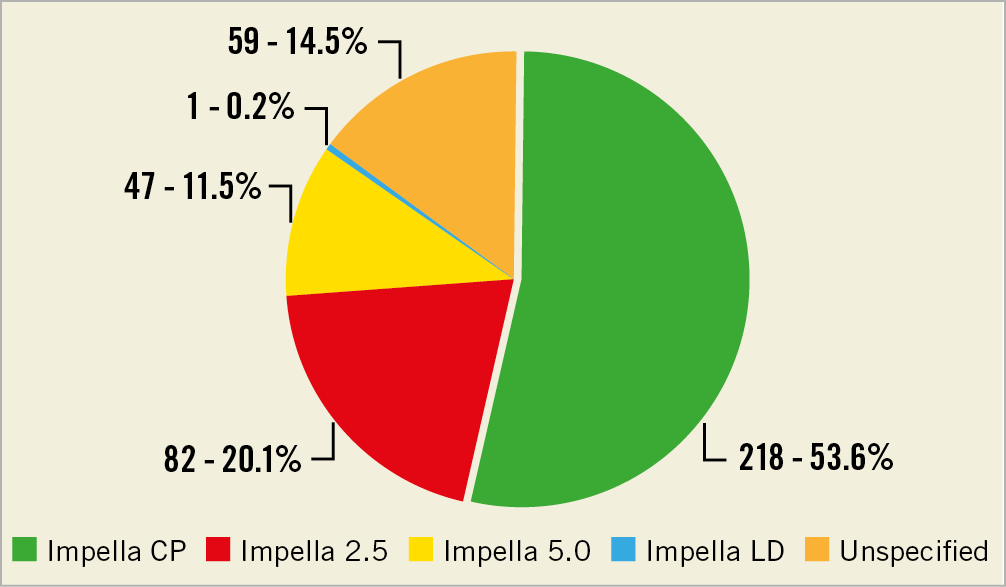
Figure 1. Adverse events stratified by the different Impella device types.
The most commonly reported complication was bleeding, which represented 38% of MAUDE reports, of which 70.3% required transfusion of packed red blood cells. Significant vascular complications, including dissection and perforation, were documented in 67 reports (16.4%). Of the 407 MAUDE reports, 168 (41.2%) confirmed that the Impella device was returned to the manufacturer for analysis; the remaining were either discarded or held at the facility at the time of this analysis. The most commonly reported failure mode was failure of the device components, noted in 29.9% of reports. Device malfunction and device separation were reported in 70 (17.2%) and 39 (9.5%) reports, respectively. Supplementary Table 2 and Supplementary Table 3 summarise proportions of reported complications and failure modes. Figure 2 outlines the temporal trends for annual reporting of the adverse events related to different Impella devices.
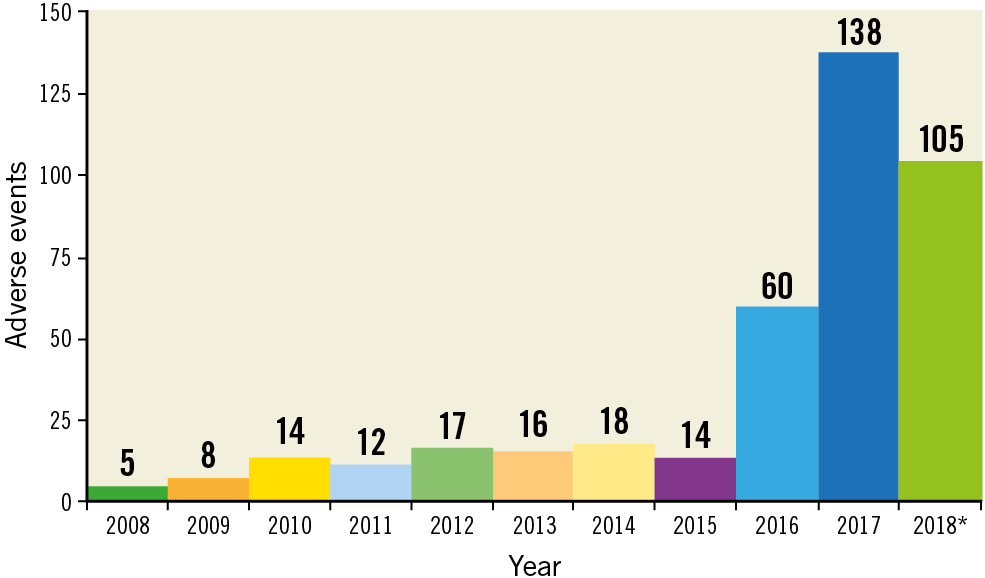
Figure 2. Temporal trends for annual reporting of the adverse events related to different Impella devices. *Incomplete year – data reported to 31 August 2018.
Discussion
The salient findings of our analysis are: a) the highest number of device-related adverse events was for the Impella CP, b) the majority of patients received the Impella for HRPCI, c) the most commonly reported complications were bleeding requiring blood transfusion and vascular complications, d) the most commonly reported failure modes included mechanical damage of the device components and device malfunction, and e) annual reporting trends for Impella MDRs show an upward trajectory, probably reflecting increased use of Impella devices.
Data regarding the incidence of adverse events related to Impella devices are scarce. In the pivotal trials of the Impella 2.5 in HRPCI patients, the incidence of major adverse events was 20% in PROTECT I, 8% in USpella, and 35.1% in PROTECT II3. In the Europella registry, bleeding and vascular complication rates were reported at 6.2% and 4%, respectively3. In the USpella registry patients developing AMI with cardiogenic shock and receiving Impella 2.5, reported complication rates were bleeding requiring transfusion (17.5%), vascular complication with surgical repair (9.7%), renal failure (18.1%), and haemolysis (10.3%)4. A prospective analysis of an Impella database reported vascular complication rates of 17%, with amputation rates of 4.4%5. It is important to understand that our analysis provides insights into the mechanism of device-related complications but cannot verify causality, neither does it provide information regarding incidence rates for individual complications. The total number of Impella units implanted during the study period remains unknown; however, the Impella Quality database reports this number to exceed 46,000 between 2009 and 20176.
In our analysis, the majority of patients received the Impella device for HRPCI. Use of the Impella is feasible in these patients3; however, limb ischaemia, bleeding requiring transfusion, and vascular access-related complications are important potential complications to consider1,5. pLVADs have a clear role in select cases of HRPCI; however, it is not clear that these devices should be the standard of care for all HRPCIs. Rather, pLVADs should remain a standby adjunctive therapy in many cases. More data are needed to define better the patient population that will derive the greatest benefit from these devices while minimising the risk of complications. The onus falls on both the clinicians to individualise patient care on a case-by-case basis and the cardiovascular device industry to continue improving device technology to achieve optimal outcomes. Newer device iterations, with smaller calibre sheath dimensions for example, may help to mitigate many of these adverse events.
Limitations
Without on-site evaluation, causality attribution cannot be established between the Impella device and adverse events. A minority of the devices were returned to the manufacturers for evaluation following the procedure, preventing a complete analysis of failure modes. Incidence rates for each complication could not be determined because of the lack of a denominator. Some general limitations of the MAUDE database include the fact that adverse events may be reported by users and manufacturers, leading to duplicate reports. Since the reporting is mostly voluntary, an unknown number of complications remains unregistered. Adverse events caused by clinician error may be underreported or inappropriately attributed to device failure. The database may contain incomplete and unverified data.
Conclusions
Analysis of the MAUDE database demonstrates that, in real-world practice, Impella devices are associated with important complications. Judicious use, appropriate patient selection, and operator experience can all help to mitigate these complications.
|
Impact on daily practice The management of patients requiring mechanical haemodynamic support is rapidly evolving, and there is a need for continued surveillance of safety profiles, patient outcomes, and failure modes for pLVADs. The MAUDE database serves as an important platform for both clinicians and manufacturers to improve device performance and optimise clinical outcomes. |
Conflict of interest statement
R. Waksman reports being an Advisory Board/Board Member and consultant for Abbott Vascular, Amgen, Pi-Cardia Ltd, CardioSet, Medtronic, Philips (Volcano), and Boston Scientific Corp., a consultant for Biosensors and Biotronik, receiving grant support from Abbott Vascular, AstraZeneca, Biosensors, Biotronik, Chiesi, and Boston Scientific Corp., being on the speaker’s bureau of AstraZeneca and Chiesi, and being an investor in MedAlliance. T. Rogers is a consultant and proctor for Medtronic, and a proctor for Edwards Lifesciences. The other authors have no conflicts of interest to declare.
Supplementary data
Supplementary Table 1. Different indications for Impella placement among reports submitted to the MAUDE database.
Supplementary Table 2. Summary of complications among reports submitted to the MAUDE database.
Supplementary Table 3. Commonly reported proportions of failure modes for Impella.
