Abstract
Aims: To demonstrate the safety, performance and efficacy of the Endeavor™ Resolute™ stent in the treatment of patients with multiple-vessel as well as single-vessel coronary artery disease but where only one lesion per patient was treated.
Methods and results: 130 patients were treated with the Endeavor™ Resolute™ stent. Of these patients, 30 consented to a 4 month follow-up evaluation by quantitative coronary angiography (QCA) and intravascular ultrasound (IVUS) measurements, and 100 consented to a 9 month follow-up evaluation by QCA. Only lesions with diameter stenoses > 50% and with lengths > 14 mm and < 27 mm in vessels with reference diameters > 2.5 mm to < 3.5 mm were included. The device success rate as 99.2%, and the procedure success rate was 96.2%. The mean lesion length was 15.56±6.27 mm. The overall 30 day incidence of major adverse cardiac events (MACE) was 3.3%, which consisted entirely of 5 cases of peri-procedural non-Q-wave MI. The QCA and IVUS results at 4 months for the 30 patient subgroup showed in-stent late lumen loss was 0.12±0.26 mm, and in segment late lumen loss was 0.05±0.20 mm.
Conclusions: The 4 month results in this subset of Endeavor™ Resolute™ patients demonstrated excellent procedural and device success when deployed in lesions up to 27 mm of length. The Endeavor™ Resolute™ stent, in this subset, was associated with a low in-stent late lumen loss and a minimal amount of neointimal hyperplastic in-growth.
Introduction
The use of drug-eluting stents (DES) in place of bare metal stents in percutaneous coronary interventions has significantly reduced the rates of clinical and angiographic restenosis1-3. The revascularisation benefit with DES, however, is attenuated in the presence of high-risk lesion and patient cohorts. Such groups include patients with diabetes4, patients with diffuse or multivessel disease, patients with chronic renal failure5, patients with left main disease or ostial disease, and patients who present with chronic total occlusions6.
The Endeavor™ DES system (Medtronic Vascular, Santa Rosa, CA, USA) combines the antiproliferative agent, zotarolimus, with a biomimetic phosphorylcholine drug carrier and a low-profile, thin-strut, cobalt-chromium alloy stent (the Driver™ stent, Medtronic Vascular, Santa Rosa, CA, USA). To date, three randomised clinical trials with active controls have examined the safety and efficacy of the Endeavor™ stent for the treatment of patients with symptomatic ischaemic heart disease due to de novo stenotic lesions in native coronary arteries. Additional trials, both registry and randomised are under way or planned7. Results have consistently demonstrated low rates of angiographic restenosis and repeat revascularisation with the Endeavor™ stent, despite varied trial design and physician practices8-10. The evidence also suggests a superior safety profile. In a pooled analysis of 1,318 patients using protocol definitions for stent thrombosis, treatment with the Endeavor™ stent was associated with very low rates of early (0.3%, 4 patients), and no late thrombosis events11.
Endeavor™ Resolute™ is a next-generation Endeavor™ stent system now undergoing human clinical evaluation. The Endeavor™ Resolute™ DES is designed to match the efficacy and safety of the original Endeavor™ stent, while improving the clinical outcomes in more complex lesion subsets. Like the original Endeavor™ stent, the Endeavor™ Resolute™ consists of the antiproliferative agent zotarolimus and the low-profile, thin-strut Driver™ bare metal stent platform. However, instead of the phosphorylcholine coating of the original Endeavor™ stent, the Endeavor™ Resolute™ stent employs a new proprietary polymer coating to enable extended drug elution to match the delayed healing times of more complex lesions and to combat the sustained stimulus to the proliferative response in more difficult patient subsets.
The new polymer coating of the Endeavor™ Resolute™ stent is based on the BioLinx™ polymer system consisting of a unique blend of three different polymers: (1) the hydrophobic C10 polymer, which aids in the control of drug release; (2) the hydrophilic C19 polymer, which supports biocompatibility; and (3) polyvinyl pyrrolidinone, which increases the initial drug burst and enhances the elution rate. Like the phosphorylcholine coating of the original Endeavor™ stent, the new polymer coating of the Endeavor™ Resolute™ highly is biocompatible. The hydrophilic surface mimics the body’s biological chemistry, thereby reducing the risk of an inflammatory response. Autopsy examinations have identified polymer hypersensitivity as a possible risk factor for stent-thrombosis deaths associated with first-generation sirolimus- and paclitaxel-eluting stents12,13. In a study comparing the Endeavor™ Resolute™ stent, including the new polymer coating, with the Driver™ bare metal stent in porcine coronary arteries, there were no significant inflammatory differences between the cohorts at 28 days, and arterial healing in the presence of the BioLinx™ polymer was rapid and complete14. In another study comparing the original Endeavor™ stent and the Endeavor™ Resolute™ stent in porcine coronary arteries, biocompatibility was similar out to 180 days14.
The unique coating of the Endeavor™ Resolute™ stent enables a finer control of the rate of drug elution. Although the zotarolimus dose is identical for both the original Endeavor™ stent and the Endeavor™ Resolute stent – 1.6 µg per mm2 of stent surface – the elution is slower with the Resolute. In the porcine model, the Resolute stent elutes 85% of its zotarolimus content into tissue during the first 60 days post-procedure, and the remainder of the drug is completely eluted by 180 days14 (see Figure 1).
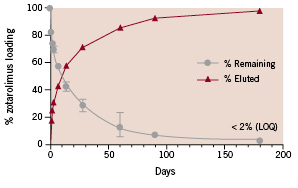
Figure 1. The in vivo elution of the Endeavor™ Resolute™ zotarolimus-eluting stent system. The Endeavor™ Resolute™ stent elutes > 85% of zotarolimus during by the first 60 days post-procedure, and the remainder of the drug is completely eluted at 180 days. From Carter et al.
The Endeavor™ Resolute™ human study of the Endeavor™ Resolute™ stent
The RESOLUTE trial is a prospective, multicentre, non-randomised, single-arm study of the use of the Endeavor™ Resolute™ stent to treat 130 patients with symptomatic ischaemic heart disease attributable to native coronary artery stenosis amenable to treatment by percutaneous stenting. Of these patients, 30 consented to a 4-month follow-up evaluation by quantitative coronary angiography (QCA) and intravascular ultrasound (IVUS) measurements, and 100 consented to a 9-month follow-up evaluation by QCA. A sample size of 130 subjects was deemed sufficient to provide reasonable confidence in the estimates of safety and efficacy generated by this study. An additional 9 patients were enrolled at 4 sites to complete the pK subset with timing of the enrolment not allowing incorporation of the data for this report.
Patients with multiple-vessel as well as single-vessel coronary artery disease were eligible to participate in this trial, but only one lesion per patient was treated. Only lesions with diameter stenoses > 50% and with lengths > 14 mm and < 27 mm in vessels with reference diameters > 2.5 mm to < 3.5 mm were included.
Twelve sites in Australia and New Zealand participated in this study. Stents were implanted according to the following guidelines. Patients received aspirin (at least 75 mg daily, started 24 hours before the procedure and continued indefinitely post-procedure) and clopidogrel (> 300 mg loading dose within 24 hours before the procedure and then 75 mg daily for a minimum of 6 months post-procedure). Predilatation with a balloon equal to or less than the proposed final stent length was mandatory.
The primary endpoint of the Endeavor™ Resolute™ trial is 9 month in-stent late lumen loss, defined as the difference between post-procedure minimal lumen diameter and follow-up minimal lumen diameter, as measured by QCA. This will be presented at EuroPCR in Barcelona in May, 2007, and published subsequently. Secondary endpoints include major adverse cardiac events (defined as death, myocardial infarction, emergent cardiac surgery, or repeat revascularisation of the target lesion); acute device, lesion, and procedure success; and angiographic parameters. Coronary angiograms performed at baseline and at follow-up were reviewed by an independent angiographic core laboratory (Brigham & Women’s Angiographic Core Laboratory, Boston, MA, USA). IVUS images were also examined by an independent core laboratory (Cardiovascular Core Analysis Laboratory, Stanford University, Stanford, CA, USA). The study was conducted according to the Declaration of Helsinki, the ethics committees of all sites approved the study protocol, and written informed consent was obtained from every patient.
Endeavor™ Resolute™: results at 4 months and discussion
The 4-month results from the Endeavor™ Resolute™ study indicate that the Endeavor™ Resolute™ stent is a safe device, with the potential for a very low rate of clinical events.
Baseline clinical characteristics for all 130 patients and for the 30 patients undergoing 4-month QCA and IVUS examination are reported in Table 1.
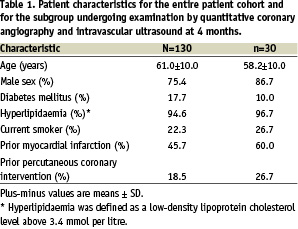
The age, sex-distribution, and risk-factor profiles of the study patients were consistent with those of a population of patients presenting with ischaemic symptoms due to a discrete de novo lesion in a single native coronary artery.
Procedural and lesion characteristics are reported in Table 2.
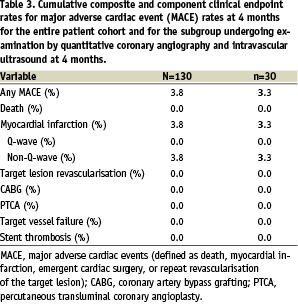
For the entire study group, the device success rate as 99.2%, and the procedure success rate was 96.2%. The mean lesion length was 15.56±6.27 mm. Clinical follow-up at 4 months for the full 130 patients is reported in Table 3.
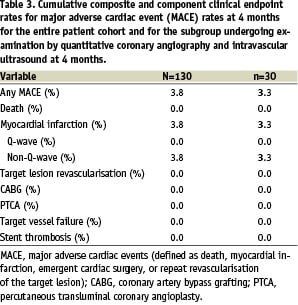
The overall 30 day incidence of major adverse cardiac events (MACE) was 3.3%, which consisted entirely of 5 cases of peri-procedural non-Q-wave MI. Three of the cases were associated with 30 mm stenting. All cases were noted to have side branch slower blood flow associated with ballooning prior to stent deployment. One patient had a prior MI with cardiac enzymes still elevated at the time of stent placement. The final patient of the five had abrupt closure and rescue of the vessel prior to stent placement.
The QCA and IVUS results at 4 months for the 30 patient subgroup undergoing these examinations are presented in Tables 4 and 5, respectively.
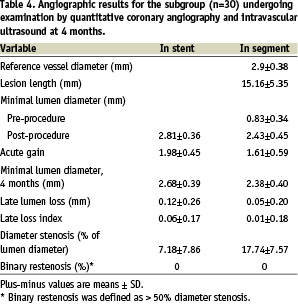
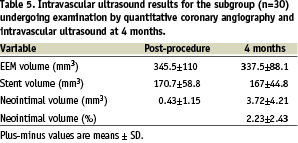
In-stent late lumen loss was 0.12±0.26 mm, and in-segment late lumen loss was 0.05±0.20 mm. This compares favourably with the 4-month follow-up data from the ENDEAVOR 1 trial, where the in-segment late lumen loss at 4 months was 0.21±0.40 mm, and in-stent late loss was 0.33±0.36 mm8. The neointimal hyperplastic volume was 0.42±1.15 mm3 post-procedure, and 3.72±4.21 mm3 at 4 months with the Endeavor™ Resolute™ stent. The percentage neointimal volume obstruction was 2.23±2.43% at 4 months. The EEM volume post procedure and at 4 months were similar, noting no significant positive or negative remodelling. Moreover, the stent volume was similar indicating no stent recoil.
Pharmacokinetic data from the Endeavor™ Resolute™ study confirms the elution profile in animals with lower Cmax and AUC, thereby increasing the drug margins of safety to over 78 fold for 48 mm stent length.
In summary, the 4-month results in this subset of Endeavor™ Resolute™ demonstrated excellent procedural and device success when deployed in lesions up to 27 mm of length. The Endeavor™ Resolute™ stent, in this subset, was associated with a low in-stent late lumen loss and a minimal amount of neointimal hyperplastic in-growth. The incidence of MACE was low at 4 months, with no cases of clinically driven target lesion revascularisation, target vessel failure, or stent thrombosis. These data in turn indicate that zotarolimus is a very potent and highly anti-restenotic agent.
Long term follow-up of this trial, as well as additional randomised trials, are planned to confirm the continued safety and efficacy of the Endeavor™ Resolute™ in complex lesion groups.
