Abstract
Aims: To demonstrate the safety, performance and efficacy of the Conor CoStar® cobalt chromium paclitaxel-eluting Stent System in the treatment of de novo coronary lesions in up to two native coronary arteries.
Methods and results: Two groups of patients were treated with the CoStar® stent in a prospective multicentre two arm registry; 145 patients received 194 stents releasing 10 µgm of paclitaxel over 30 days (Arm 1), and 137 patients were treated with 158 stents releasing 30 µgm of paclitaxel over 30 days (Arm 2). Baseline demographics were well matched. Overall device success was 98.2%. Intention-to-treat in-segment late loss at six months was 0.09±0.40 mm in Arm 1, and 0.22±0.46 mm in Arm 2 (p=0.015), with an in-stent late loss of 0.28±0.42 mm and 0.40±0.48 mm in Arm 1 and Arm 2 respectively (p=0.028), with no deleterious edge effect in either group. Cumulative MACE at 12 months in Arm 1 was 8.3%, with a stent thrombosis rate of 0.7%, and in Arm 2, 10.2% and 1.4% respectively (MACE: p=0.682). Hierarchical target lesion revascularisation (TLR) at 12 months was 2.8% in Arm 1 and 3.4% in Arm 2 respectively (p=0.759).
Conclusions: The Conor CoStar® cobalt chromium stent system is highly deliverable and when loaded with low dose slow release paclitaxel (10 µgm released over 30 days), the stent appears safe and efficacious as judged by angiographic and clinical variables assessed at six and 12 months respectively.
Introduction
Drug eluting stents loaded with either paclitaxel1-6 or sirolimus7-9 have been shown to reduce restenosis and the requirement for clinical or angiographically driven target vessel revascularisation (TLR). There is however no evidence that they reduce the other determinants of major adverse cardiac events (MACE), namely cardiac death or myocardial infarction10. In fact, there is concern that there may be an associated increase in procedural MACE compared with bare metal stents as reported in the paclitaxel eluting limbs of both the TAXUS V4 and TAXUS VI5 randomised trials. It is postulated that this increase in MACE may be due to side branch compromise by thicker polymer coated stent struts, transient platelet or thrombus deposition in association with drug elution, or paclitaxel induced spasm4. In that the polymer may be the culprit, it is logical to both reduce the absolute amount of polymer per stent, without compromising drug delivery, and to investigate the role of a bioresorbable polymer delivery system which may have a more favourable side effect profile. Furthermore, by varying the release kinetics of paclitaxel, it may be possible to determine the minimally effective dose of drug released over an optimum time-frame.
This is the first report from the EuroSTAR trial which has assessed the safety and efficacy of a unique drug delivery system with two doses of slow release paclitaxel released delivered from a low profile cobalt-chromium stent platform.
Methods
CoStar® stent system
The CoStar® stent is made from cobalt chromium steel alloy and elutes the antiproliferative drug paclitaxel via holes or wells located in the stent struts. The stent is pre-mounted on a sterile rapid exchange balloon catheter and incorporates a design feature called a ductile hinge which allows the stent to expand in a way that prevents strut and well deformation. Elution of the compound paclitaxel from the wells is mediated by the erosion of a bioresorbable polylactide-co-glycolide (PLGA) polymer matrix.
In the EuroSTAR trial two consecutive cohorts of patients were treated with CoStar® stents loaded with two different paclitaxel dose regimens. The first 145 patients (Arm 1) were treated with 10 µgm* paclitaxel eluted over 30 days (in vitro) from the mural side of the stent; the subsequent 137 patients (Arm 2) were treated with 30 µgm* paclitaxel eluted over 30 days (in vitro) from the mural side of the stent (*doses relate to a single 2.5 mm diameter, 17 mm length CoStar® stent).
Patient selection
1. Following written informed consent, patients scheduled for elective percutaneous coronary intervention were included. They had symptoms of stable or unstable angina (CCS > Class I), or a positive functional test for myocardial ischaemia. Up to two native de novo coronary lesions could be treated in multiple vessels with a reference vessel diameter of 2.5-3.5 mm and a lesion length of < 25 mm with TIMI flow > Grade 1.
2. Patients were excluded if left ventricular systolic function was poor (ejection fraction < 30%), there was visible intraluminal thrombus or evolving myocardial infarction. Bifurcation stenting with involvement of a side-branch > 2 mm diameter was not permitted. Drug hypersensitivity, recent stroke, peptic ulceration, or gastrointestinal haemorrhage were also contraindications.
Study procedure
The study was approved by the local Medical Ethics Committee in each of the investigation sites. Patients who met the study entry criteria were treated with the appropriate CoStar® stents according to lesion and vessel morphology depending on whether they were in Arm 1 or Arm 2 of the trial (see above) (Figure 1).
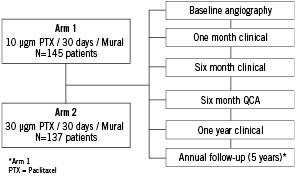
Figure 1. EuroSTAR trial: study design and patient flow.
Patients were pre-treated with aspirin > 80 mg and clopidogrel 300 mg (at least two hours prior to the procedure). A sufficient dose of intravenous unfractionated heparin was administered to maintain the ACT > 250 seconds. Intracoronary nitrates (GTN or ISDN) were given before recording the baseline and final angiograms. Percutaneous intervention was undertaken using standard techniques; CoStar® stents were implanted according to the instructions-for-use. Direct stenting was permitted in the protocol according to physician preference. Angiographic images were recorded according to the protocol of the angiographic core laboratory. A pre-discharge electrocardiogram was obtained 18-24 hours after stent placement, and serial cardiac enzyme estimations (CK and CK.MB) every 8±2 hours post procedure (x3). Aspirin > 80 mg daily and Clopidogrel 75 mg daily were continued for a minimum of six months after the procedure. Glycoprotein IIb/IIIa inhibitors were used at the discretion of the physician. Clinical follow-up was mandated at one, six, and 12 months for all patients, and annually for five years in patients treated in the Arm I group only. Coronary angiography was repeated at six months in both arms.
Data management
Site monitoring was undertaken by an independent organisation (MedPass International, Paris, France); the investigators had unrestricted access to the data. An independent core angiographic laboratory (Cardialysis BV, Rotterdam, The Netherlands) analysed the angiograms. All major adverse cardiac events were reviewed and adjudicated by an independent Clinical Events Committee, and a Data Monitoring Committee periodically reviewed blinded safety data.
Study endpoints
The primary objective of the EuroSTAR trial was to evaluate the safety and performance of the Conor paclitaxel-eluting CoStar® stent in reducing angiographic restenosis when used in de novo native coronary artery lesions. The primary endpoint was angiographic late loss within the stent at six months post procedure as measured by core laboratory quantitative coronary analysis (QCA).
Secondary endpoints included primary device, lesion and procedural success, together with major adverse cardiac events (MACE) at 30 days, 6 and 12 months. MACE was defined as cardiac death, myocardial infarction (Q- or non-Q-wave), emergent cardiac bypass surgery, and clinically driven target lesion revascularisation (TLR). Angiographic variables include in-stent and in-segment (stent±5 mm) binary restenosis, minimum lumen diameter (MLD), and late loss at six months. Clinical efficacy is defined by target lesion revascularisation (TLR) and target vessel revascularisation (TVR) rates at six months.
Statistical analysis
The primary analysis sample included all patients who received the CoStar® stent per protocol and who complete clinical and angiographic follow-up. Secondary safety analysis was conducted on all patients who are enrolled in the study. Descriptive statistics were used to summarise the data by treatment group. Categorical variables are presented as counts and incidence rates. Continuous variables are presented as mean, standard deviation, median, minimum, maximum and number of observations. The event variables (including MACE) are summarised as time-to-event variables and presented using the Kaplan-Meier method.
Results
Baseline demographics are presented in Table 1.
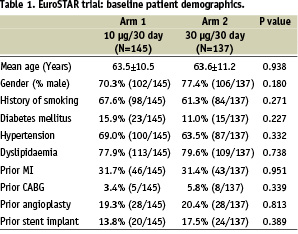
The two arms were well matched with features typical of patients undergoing percutaneous coronary intervention in a ‘first-in-man’ (FIM) study; the low incidence of diabetes mellitus reflected the per protocol lesion characteristics of the trial. In Arm 1, 176 lesions were treated with 194 stents in 144 patients; in Arm 2, 145 lesions were treated with 158 stents in 134 patients (Table 2).
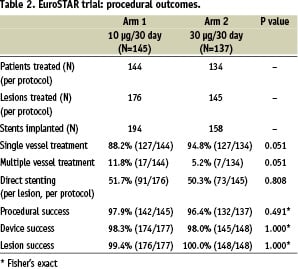
By chance, more patients were treated with multivessel disease in Arm 1 (Arm 1; 17/144, 11.8%. Arm 2; 7/134, 5.2%; p=0.051). Approximately half the patients were treated by direct stenting (without balloon pre-dilatation) in each group. Lesion, device and procedural success was high in each group.
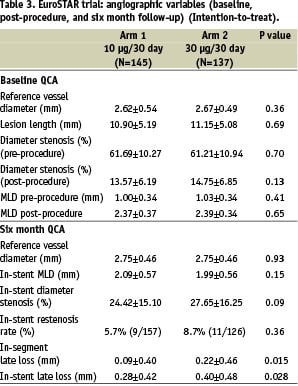
Lesions were well matched for baseline QCA between the groups in intent-to-treat analysis. The primary endpoint of in-stent late loss at six months was significantly lower in Arm 1 than in Arm 2 (0.28±0.42 mm vs. 0.40±0.48 mm respectively; p=0.028). In-segment late loss was also less in Arm 1 than Arm 2 (0.09±0.40 vs. 0.22±0.46 mm respectively; p=0.015). There was no deleterious edge effect (Figure 2).
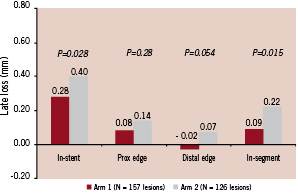
Figure 2. EuroSTAR Trial: late loss (six months).
Major adverse cardiac events (MACE) at 30 days, six and 12 months are documented in Table 4. There were no significant differences in MACE between Arm I and Arm 2 at 30 days, although there was a trend towards a higher MACE in Arm 2 compared with Arm 1 (5.8% vs. 1.4% respectively, p=0.055); by 12 months cumulative MACE in Arm 1 was 8.3% (12/145), and in Arm 2, 10.2% (14/137) (p=0.682). Hierarchical TLR at 12 months was 2.8% (5/176 lesions) and 3.4% (5/145) in Arm 1 and 2 respectively (p=0.759). The denominator for patient compliance was based on the number of patients eligible for follow-up (see Tables 3 and 4).
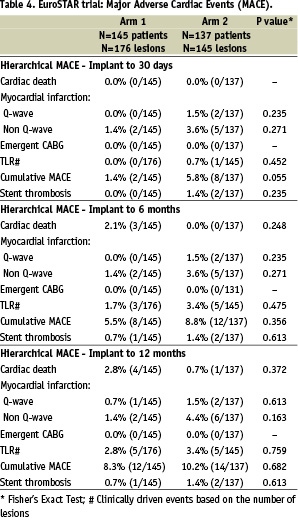
There were five cardiac deaths in the study during the 12 month follow-up period, 4/145 (2.8%) in Arm 1 and 1/137 (0.7%) in Arm 2. In addition there was one non-cardiac death in Arm 2; thus, all-cause mortality was 4/145 (2.8%) in Arm 1 and 2/137 (1.5%) in Arm 2. In Arm 1, one patient died of heart failure on day 53, one patient died suddenly of an LAD stent thrombosis on day 53, one patient died suddenly on day 195 with a patent stent at autopsy, and one patient died of left ventricular failure on day 378 (within the trial time window of 365±30 days). In Arm 2, the single cardiac death occurred on day 273 from an unknown cause, but was presumed to be cardiac in the analysis. This patient was initially compliant with dual anti-platelet therapy during the first month, and then subsequently required treatment to the left main stem with a non-study (TAXUS™) stent at six month angiographic follow-up. Three stent thromboses occurred in the study (3/282, 1.06%), one in Arm I and two in Arm 2. The single stent thrombosis in Arm 1 occurred on day 53 and resulted in one of the cardiac deaths (above), the first stent thrombosis in Arm 2 resulted in a non Q-wave myocardial infarction on day four. The second stent thrombosis in Arm 2 occurred in a patient who the CEC determined had died a non-cardiac death as a consequence of a gastrointestinal haemorrhage and oesophageal rupture on day 9 post procedure.
Discussion
The uptake of drug eluting stent technology in Europe varies from 9% to 88% (mean 52%) between countries11; the limitation often reflecting a cash constrained health care system, rather than the reluctance of the operator to use the device. Enthusiasm for drug eluting stents mirrors the perceived efficacy in reducing the requirement for target lesion revascularisation; contemporary day-to-day clinical practice clearly indicates that it is uncommon for a patient to return with symptomatic restenosis within a drug eluting stent. Increasing experience with drug eluting stents has however resulted in concern over late stent thrombosis12,13 which may be more common with stents that elute paclitaxel14,15, although this finding has been reported inconsistently16. The causes of late stent thrombosis remain unclear but it is likely that partial or incomplete endothelialisation due to drug toxicity, a reaction to the polymer, or the premature discontinuation of anti-platelet therapy may all play a part. Thus, efforts to optimise the choice of drug, the dose, the release kinetics, the polymer and the stent delivery platform continue.
Optimal drug delivery requires a stent platform with favourable performance characteristics that enables the drug to reach the target lesion. In the EuroSTAR trial we report for the first time the use of the CoStar® cobalt chromium (L605) stent which combines a low profile highly deliverable stent with excellent fluoroscopic visualisation. The strut thickness of 0.09 mm results in a crossing profile of 0.98 mm for a 2.5 mm diameter stent, the lowest profile presently available for a drug eluting stent. The CoStar® stent utilises reservoir drug delivery technology which allows precise, directional and variable dosing without the need for a surface polymer coating, thereby reducing the stent profile and the overall polymer dose. Individual drug delivery wells are filled with a bioresorbable polylactide-co-glycolide (PLGA) polymer and drug matrix that does not protrude from the well, thus it does not impact on the profile of the device. The polymer matrix has been shown to be completely resorbed within 180 days in a porcine model (unpublished data on file, Conor Medsystems), with the expectation that the CoStar® stent will have a similar late safety profile to a bare metal stent following complete elution of the drug and resorption of the polymer.
The cellular action of paclitaxel is complex, principally involving the inhibition of cell division by binding to ß-tubulin, stabilising microtubular dynamics, inhibiting microtubular assembly, and disrupting the equilibrium between tubulin dimers and tubulin polymers. Although paclitaxel has been shown to have a beneficial effect on vessel injury and neointimal proliferation in animal models17-19, high dose paclitaxel may inhibit re-endothelialisation and cause toxicity both in animal models20 and in man21.
In the porcine model, it is apparent that paclitaxel has a narrow therapeutic window and that modification in the drug dose or the release kinetics may have a significant effect on the tissue response to stent injury22. This observation was confirmed in man in the PISCES study using a stainless steel MedStent™ with reservoirs loaded with either 10 or 30 µgm of paclitaxel released over 5, 10 or 30 days, in either a luminal or abluminal direction23,24. It was clear that the slow release (30 days) formulation was superior in terms of clinical outcome, and although the 30 µgm dose had lower angiographic late loss and a reduced neointimal volume by IVUS at one year compared with the 10 µgm dose, this did not reach statistical significance. Of the six groups studied in PISCES, the two that demonstrated the best efficacy formed the basis of the EuroSTAR trial, namely 10 µgm of paclitaxel released over 30 days (Arm 1), and 30 µgm of paclitaxel released over 30 days (Arm 2).
Durable polymers allow predictable drug release as well as the ability to manipulate dose release kinetics, but the polymers themselves are known to induce an inflammatory reaction25,26 and have been implicated as a potential cause of early MACE in the TAXUS V trial4. Attempts to coat stents with paclitaxel without a polymer have met with varied success. When paclitaxel is applied directly to the abluminal surface of a stent a clear dose response curve has been demonstrated in both the ASPECT trial27, using the Supra-G™ stent with paclitaxel doses of 1.3 and 3.1 µgm/mm2, and in the ELUTES trial28 using the V-Flex Plus™ stent with paclitaxel doses of 0.2, 0.7, 1.4 and 2.7 µgm/mm2. Both these trials confirmed that higher doses of paclitaxel were more effective at inhibiting neointimal proliferation without any deleterious safety concerns in this dose range. In contrast, the DELIVER trial29, which used the Achieve stent (a Multilink Penta® Stent coated with 3.0 µgm/mm2 paclitaxel using the Cook non-polymeric coating process), failed to reach the primary end point of target vessel failure. Although there was a significant reduction in angiographic in-stent late loss (0.81 mm vs. 0.98 mm; p=0.003), this was not reflected in clinical efficacy.
The EuroSTAR trial has confirmed that the CoStar® cobalt chromium stent system is highly deliverable with an overall device success rate of 98.2%; furthermore, a direct stenting strategy was used successfully in more than half the lesions. The primary endpoint of angiographic in-stent late loss at six months (Table 3) was significantly lower in Arm I than in Arm 2 (0.28±0.42 mm and 0.40±0.48 mm respectively; p=0.028), suggesting that the lower dose of paclitaxel (10 µgm) used in Arm 1 has been associated with less toxicity than the higher dose (30 µgm) used in Arm 2. The in-stent late loss of 0.28 mm in Arm 1 compares favourably with the in-stent late loss seen in the TAXUS II - VI polymer-based paclitaxel trials which varied from 0.30 - 0.49 mm1-6. Furthermore, there was no deleterious edge effect observed in the EuroSTAR trial in either group (Figure 3).
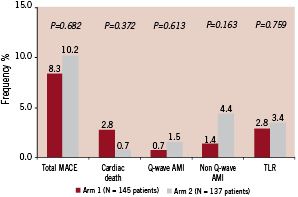
Figure 3. EuroSTAR trial: clinical events (12 months). Hierarchical MACE.
Hierarchical MACE at 12 months was 8.3% in Arm 1 and 10.2% in Arm 2 (p=0.682), with a low 12 month rate of hierarchical target lesion revascularisation of 2.8% and 3.4% respectively (p=0.759) (Table 4); both variables trended lower in the 10 µgm group (Arm 1). All cases of target lesion revascularisation were treated by repeat percutaneous intervention rather than coronary artery bypass grafting. This trial is underpowered to make any definitive conclusions in relation to stent thrombosis. One stent thrombosis occurred in Arm I (0.7%) and two stent thromboses occurred in Arm II (1.4%). A 2.5 mm x 16 mm CoStar® stent implanted in the mid LAD occluded on day four (Arm 2) resulting in a non Q-wave myocardial infarct (peak CK 2000 iU/l); the stent was successfully recanalised and additional disease was treated in the postero-lateral circumflex. A second stent thrombosis occurred in a patient on day 53 (Arm 1) resulting in sudden death, autopsy confirming a thrombosis in a 3.5 mm x 17 mm CoStar® stent in the LAD, with an additional (untreated) occlusion of the left circumflex. Both patients with stent thrombosis were assumed to be taking dual anti-platelet therapy at the time of the event. After the cessation of dual antiplatelet therapy at six months, there were no additional stent thromboses between six and 12 months when patients were treated with aspirin alone.
Conclusions
The Conor CoStar® cobalt chromium stent system is highly deliverable and when loaded with low dose slow release paclitaxel (10 µgm released over 30 days), the stent appears safe and efficacious as judged by angiographic and clinical variables assessed at six and 12 months respectively in the EuroSTAR trial. The utility of reservoir drug delivery has yet to be fully exploited. Not only can this technology allow directional elution (luminal vs. abluminal) from a single reservoir, but individual wells can be loaded with drug doses with variable kinetics, or alternatively, different drugs with diverse properties. Combinations of drugs in lower dose may target different intra-cellular pathways and be associated with less toxicity. Based on favourable data from a porcine model30, the GENESIS trial which combines the abluminal elution of paclitaxel and pimecrolimus in adjacent wells is now under way.
Study limitations
The EuroSTAR trial is typical of a ‘First-in-Man’ study with relatively simple lesions and a low incidence of diabetes; the results may not therefore reflect ‘real world’ practice.
Clinical events committee
Alan C. Yeung (Chairman), Palo Alto, USA; Frederick St. Goar, Palo Alto, USA; David P. Lee, Palo Alto, USA.
Data safety and monitoring board
James D. Joye (Chairman), Mountain View, USA; Tomoaki Hinohara, Redwood City, USA; Jonathan Tobis, Los Angeles, USA.
Participating investigators & centres
Keith D. Dawkins (PI): Southampton University Hospital, Southampton, United Kingdom
Antonio Colombo (PI): Ospedale San Raffaele, Milan, Italy
Stefan Verheye: AZ Middelheim Hospital, Antwerpen, Belgium
Helmut Schühlen: Medizinische Klinik rechts der Isar, Technische Univ Munich, Germany
Joseph Dens: Universitair Ziekenhuis, Leuven, Belgium
Harald Mudra: Städtisches Klinikum München GmbH, Klinikum Neuperlach, Munich, Germany
Wolfgang Rutsch: Universitatsklinikum Charite Mitte Humboldt Univ, Berlin, Germany
Pieter R. Stella: Universitair Medisch Centrum, Utrecht, The Netherlands
Carlo DiMario: Royal Brompton Hospital, London, United Kingdom
Martyn Thomas: King’s College Hospital, London, United Kingdom
Peter den Heijer: Amphia Hospital de Klokkenberg, Breda, The Netherlands
Francois Schiele: CHU Jean Minjoz, Besançon, France
Patrick Serruys: Thoraxcentrum, Erasmus University, Rotterdam, The Netherlands
Dougal McClean: Christchurch Hospital, Christchurch, New Zealand
Victor Legrand: CHU Sart Tilman, Liege, Belgium
Maarteen J Suttorp: Sichting Sint Antonius Ziekenhuis, Nieuwegein, The Netherlands
Gilles Grollier: CHU Cote de Nacre, Caen, France
Charles Ilsley: Harefield Hospital, Harefield, United Kingdom
