Introduction
The feasibility of transcatheter aortic valve implantation using metallic stent-based tissue valves in patients at high-risk for surgical aortic valve replacement has recently been demonstrated.1-4 Although initial clinical results have been encouraging, these first generation devices are neither repositionable nor retrievable after deployment. To address these concerns, a novel, inflatable prosthetic heart valve, which is initially percutaneously deployed in the collapsed configuration, and subsequently inflated to an operative configuration was conceived in the early 1990’s.5,6 The concept of an inflatable cuff tissue valve was further developed and validated using extensive preclinical testing by Direct Flow Medical, Inc. (Santa Rosa, CA, USA) and is now being evaluated in a European-based clinical trial. This device facilitates consistent and accurate placement of the device by utilising a step-by-step positioning method. Furthermore, if the clinical or haemodynamic results warrant, the device is completely retrievable via an integral recovery system through the introducer sheath. We herein describe the extensive preclinical development and testing prior to first-in-human implants.
Device description
The Direct Flow Medical percutaneous aortic valve (PAV) system is comprised of three key components: the bovine pericardial tissue heart valve, the sheathed delivery system with integral recovery system, and solidifying inflation medium that forms the support structure.
Bovine pericardial tissue heart valve
The Direct Flow Medical PAV is designed to have the durability of a surgical valve and includes an anti-calcification treatment of the bovine pericardial leaflets. The tri-leaflet tissue valve is attached to an inflatable Dacron fabric cuff which conforms to the native annulus to create a tight seal, minimising paravalvular leak. The implant is designed in an hourglass shape, with independently inflatable ventricular and aortic rings, which encircle and capture the native valve annulus ensuring positive anchoring of the device and to provide the largest effective orifice area. The implant is made functional initially by inflation with saline and contrast (50/50 mixture), allowing fluoroscopic visualisation of the ring elements. At the end of the procedure the saline contrast mixture is exchanged for a solidifying inflation medium that hardens to form the permanent support structure.
The PAV is available in multiple sizes. Two sizes (23 and 25 mm) are available for use in the initial European clinical trial (Figure 1).
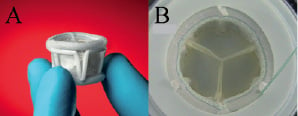
Figure 1. Direct Flow Medical percutaneous aortic valve. (A) Side view of current generation devices showing aortic and ventricular inflation rings. (B) Top view of device in hydrodynamic tester during simulated diastole.
These devices have aortic ring outer diameters of 27 and 29 mm, total valve heights of 16 and 17 mm and calculated effective orifice areas of 1.9 and 2.6 cm2, respectively (hydrodynamic testing performed at 5 L/min, 120 mmHg and 70 bpm).
Solidifying inflation medium
The Direct Flow Medical PAV system utilises contrast and saline to initially expand the valve. When the position is acceptable, the contrast and saline are exchanged out with a proprietary inflation medium (IM) while maintaining pressure to keep the valve expanded. The IM is a biocompatible, two component liquid containing a water soluble epoxy and a radiopacifier. This unique technology creates the support structure in situ.
The IM is currently provided in two syringes that are mixed extra-corporally by the physician and delivered to the implant via the delivery system. The IM displaces the contrast and saline from the pressurised Dacron cuff. After the exchange, the IM gels in minutes and achieves 95% of its final hardness within hours. After the IM exchange is complete, and at all times during the curing process, 8 atmospheres (120 psi) of pressure is maintained in the Dacron cuff. The end result is a strong, durable polymeric support structure which is formed in situ and permanently fixes the implant in the native annulus. The IM in the liquid form is designed to be water soluble and non-embolic if released into the blood stream.
Delivery and recovery systems
The Direct Flow Medical delivery system is a 15 Fr multi-lumen catheter with an outer sheath at the distal end that houses the implant. The distal portion of the outer sheath is currently 22 Fr. The catheter contains three position / fill lumens (PFLs) which are attached to the implant. Two of these PFLs are used to inflate and deflate the cuff and all three are used to position and align the implant precisely in the native annulus. This enables positioning and repositioning of the implant (Figure 2).
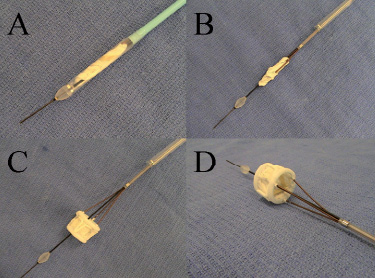
Figure 2. Direct Flow Medical delivery system. (A) Valve sheathed in delivery system. (B) Valve unsheathed prior to ring inflation. (C) Fully inflated valve pre-detachment, side view. (D) Inflated valve pre-detachment, superior view.
A handle is on the proximal end of the delivery system which contains a locking mechanism to control the PFLs and facilitate positioning of the implant. Integral to the delivery system is a recovery device which allows smooth retrieval of the Direct Flow Medical valve through a 22 Fr introducer sheath (Figure 3).
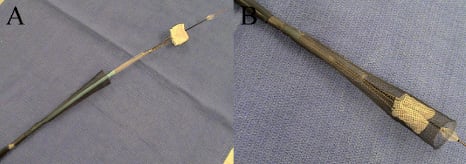
Figure 3. Direct Flow Medical recovery system. (A) Nitinol retrieval basket prior to recovery of implant. (B) Recovery system with valve retrieved and fully captured.
The entire delivery system works over a 0.035” guidewire. A smooth transition from the guidewire to the outer sheath is achieved by an atraumatic tip located at the distal end of the delivery system (Figures 2 and 3). The tip, which is maintained in the left ventricle (LV) until the PFLs are disconnected from the implant, can be used to obtain LV pressure measurements when the guidewire is retracted.
Implantation technique
The implantation procedure is shown in the supplemental video available at www.eurointervention.org. Prior to the procedure, the implant is loaded into the Direct Flow Medical delivery system (Figure 2A). Arterial access is gained percutaneously or via a surgical cut down followed by insertion of a 22 Fr sheath. Prior to device insertion, in clinical trials, balloon valvuloplasty can be performed using standard techniques to ensure adequate separation of the native aortic valve leaflets. For preclinical animal studies, the valve is implanted in the native healthy aortic or mitral annulus without predilation.
During the procedure, the constrained valve is advanced to the native valve location over a super stiff 0.035” guidewire. Once the delivery system is in the left ventricle, the outer sheath is retracted exposing the implant (Figure 2B). The initial deployment of the inflatable Dacron cuff is accomplished by injecting contrast and saline (50/50) through the PFLs . The ventricular and aortic rings are partially inflated with the entire implant located in the LV. At this point, the valve is functional and ready to be positioned in the left ventricular outflow tract. Independently the aortic ring is deflated while the ventricular ring is inflated to 8 atmospheres of pressure using standard indeflators. The device is then retracted until the ventricular ring is seated against the ventricular side of the native annulus. Because the valve function is seamlessly transferred from the native valve to the prosthetic valve, rapid pacing or cardiac support is not required during device placement. Furthermore, as the valve functions immediately upon ventricular ring inflation, there is no urgency to seat the device immediately so care can be taken to ensure precise placement.
After the ventricular ring is seated on the ventricular side of the annulus, the aortic ring is inflated to 8 atmospheres, again with a standard indeflator (Figures 2C, 2D). If the device is not in the optimal location, the device can be partially deflated and manipulated using the PFLs until optimal positioning is achieved. If a different sized implant is required, the device may be completely deflated and removed from the patient using the retrieval basket which is integral to the delivery system. Once final device function is evaluated by fluoroscopy, echocardiography and haemodynamics, the inflation media is introduced into the pressurised implant through the PFLs, displacing the contrast and saline out of the Dacron cuff and back through the proximal end of the delivery catheter. During the IM exchange which is designed to take less than five minutes the valve remains fully pressurised, in position, and competent. Following the IM exchange the implant is detached from the delivery system and the delivery system is removed from the patient. Device detach and removal takes less than five minutes.
Pre-clinical experience
Cadaver and explanted heart studies
An acute evaluation of the Direct Flow Medical PAV system was performed in perfused human cadavers and in human cadaver explanted hearts.
As part of Direct Flow Medical’s design verification program, a study was conducted to evaluate the design performance in a human cadaveric model that employs a saline-perfused heart and vascular tree (MERI Center, Memphis, TN, USA) to emulate the in vivo environment. The study evaluated the Direct Flow Medical PAV and delivery system in cadaver models and in ex vivo human hearts.
The primary focus of these studies was to evaluate the functional characteristics of the delivery system under simulated use conditions, assess the accuracy of placement of the heart valve and migration resistance, verify compatibility of the delivery system with standard accessory devices, evaluate visualisation of key product features under fluoroscopy, and confirm the implant procedure instructions. While animal models offer functioning arterial circulation, most animal vessels are too small to mirror human conditions and the anatomy of the aortic annulus is neither comparable nor diseased. Therefore, design verification tests were performed in six perfused human cadavers.
The Direct Flow Medical catheter and implant were confirmed to have excellent trackability and flexibility through the human vasculature due to its non-metallic design. Design confirmation was demonstrated with the delivery system’s ability to reach the intended location and position the Direct Flow Medical PAV into the native aortic valve. The device was placed without difficulty and the retrieval of the PAV using the recovery basket was also assessed. The PAV was placed with success and without evidence of migration and the device was fully visible under fluoroscopy (Figure 4).
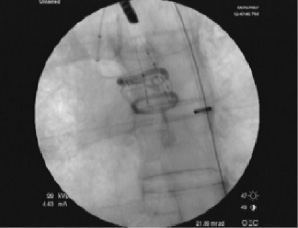
Figure 4. Direct Flow Medical valve implant in cadaveric model. The valve rings are clearly seen on fluoroscopy in appropriate position in this cadaveric model.
The Direct Flow Medical PAV and delivery system was also confirmed to be compatible with the appropriate accessory devices.
The explanted human heart studies confirmed the device’s ability to hold open severely calcified leaflets and implant resistance to pull forces in either the ventricular or aortic direction. The implant, when placed in the native aortic valve of a previously explanted cadaver heart, had an acceptable tensile strength and demonstrated sufficient migration resistance. This testing was performed in eight human cadaver hearts during the device design and pre-clinical evaluation phase of development.
Animal studies
During the development of the Direct Flow Medical PAV and delivery system over 60 animal studies were conducted. The majority of these pre-clinical studies have been acute studies performed to assess the iterative changes to the device design, evaluate the delivery system, and to study the initial deployment and performance of the PAV in vivo following IM exchange.
As part of Direct Flow Medical’s design verification program of the PAV, a series of 18 chronic animal studies were conducted in an ovine model. In these studies, the Direct Flow Medical valve was placed surgically in either the native aortic or mitral annulus due to the anatomical differences between the ovine anatomy and the human anatomy. The primary focus of the study was to evaluate the performance of the Direct Flow Medical PAV at the time of placement and to assess the biological reaction of the PAV at the time of removal. Serial explants were performed over time and evaluations were completed at one day, 30 days, 90 days and longer. The longest implant to date has been 365 days at which time the animal was electively euthanised. Follow-up echocardiography exams were performed. Gross and histopathological analyses were performed on the chronic explants. The native response to the implant and healing of the prosthesis appears to be performing as designed in either the aortic or mitral annular position. Figures 5 and 6 illustrate representative examples of excellent device healing in vivo on a chronic basis.
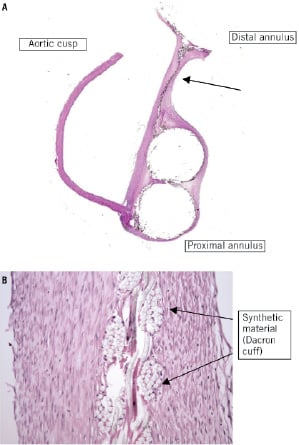
Figure 5. Valve histology. Histology slides of 80 day chronic implant in ovine model. (A) Histopathological whole mount section (H&E stained section) of the PAV implanted in the aortic position in an ovine model explanted at 80 days. (B) Medium power image of wall portion (see arrow in Figure 5A) of device showing thin pannus/fibrous tissue overgrowth of the synthetic material of the wall. Note the lack of inflammation. (H&E stained section, 10x objective)
Inflation medium biocompatibility
Integral to the design of the Direct Flow Medical PAV system is the use of contrast and saline to initially expand the valve, followed by a fluid exchange to fill the cuff with the solidifying IM that forms the device support structure in situ. Acute and chronic studies evaluating the biocompatibility of the IM were conducted per ISO requirements. Additional non-standard tests were developed and performed beyond the ISO requirements to demonstrate safety.
Due to the unique and proprietary nature of the polymeric construct of the IM several acute and chronic experiments in an ovine model to demonstrate safety of the water soluble material were performed. The method of study included injecting a prorated volume of the IM (2 ml) over a set period of time after mixing the two part material in the ascending aorta in the area of the sinus of valsalva. The entire volume of the implant and delivery system is 2 ml and the animals used in this model have approximately half the blood volume of humans. Angiography of the coronary arteries, the great vessels and the descending aorta was performed post injection and EKGs were obtained pre, post and during the injection. Full blood chemistries were performed prior to the IM injection and at one hour following the injection in the acute animal studies. In addition, the chronic ovine(s) had blood chemistries prior to the explants and termination of the study. The animals were survived for 30±7 days and observed for typical capabilities following the IM injection and recovery from anaesthesia as well as pathology and histology of the major organs at the time of sacrifice. Pre-termination coronary angiography was also performed.
The results of the standard and non-standard tests demonstrated a safe and effective design of the IM and no abnormal findings were seen with blood chemistries, EKGs, angiographic or histological results confirming the safety of the IM in vivo.
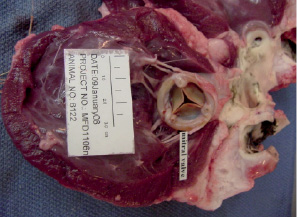
Figure 6. Gross pathology. Gross pathological section of the Direct Flow Medical PAV implanted in the mitral position of an ovine animal explanted at 365 days demonstrating excellent healing, valve function, no thrombus and minimal calcification on the pericardial leaflets or commissures.
Durability testing
Significant mechanical testing has been performed to date to develop the Direct Flow Medical PAV and validate its design. Pulsatile-flow testing is performed to optimise and verify hydrodynamic performance of the valves. All tests were performed in 0.9% saline under physiological conditions. The testing indicates that the Direct Flow Medical PAVs exceed the hydrodynamic performance requirements defined in ISO 5840 for surgical valves7.
Accelerated wear testing (AWT) in the Vivitro HiCycle testers per ISO 5840 has also been performed. The degree of damage was assessed at various intervals and was comparable to the control devices (Perimount Valve, Edwards Lifesciences, Inc., Irvine, CA, USA) used in the study (Figure 7).

Figure 7. Cycle Testing. Coaptation between T=100 million (left) and T=200 million cycles (right) in durability tester.
AWT results have been obtained beyond 300M cycles which exceeds the ISO 5840 surgical valve durability requirement. Testing was performed at 95 mmHg at test conditions described in ISO 5840. Leaflet motion was compared to video recordings made in the hydrodynamic test apparatus to ensure complete opening and closing of the valve.
In addition to the fatigue testing of the valve leaflets, extensive testing of the polymer support structure has been performed. Historically, creep deformation has been an issue for some surgical valves using a polymer support structure. Creep deformation is the tendency of a material to deform over time as a result of exposure to temperature and levels of stress below the yield strength of the material. With the Direct Flow Medical PAV, no measurable creep deformation has been detected and no fatigue fractures of the polymer support structure have been identified throughout the five year equivalent test performed to date.
Conclusion
The Direct Flow Medical PAV is functional and safe as demonstrated by extensive pre-clinical testing. Animal and cadaver testing indicate the delivery and recovery of the device can be accomplished in a series of straightforward steps. The devices have performed beyond 200M cycles in accelerated wear testing and 365 days in vivo. Results to date have demonstrated excellent safety, biocompatibility, with preserved function and long-term durability. Currently a European prospective, single arm safety and performance study is on-going for non-surgical patients with severe aortic valve disease.
Acknowledgements
The authors acknowledge the following employees at Direct Flow Medical, Inc. for their engineering and clinical research contributions: Randy Lashinski, Gordon Bishop, Do Uong, Brian Sheahan and Julie Selstrom. We acknowledge the Sutter Institute for Medical Research (Sacramento, CA, USA) and its staff for assistance in percutaneous device development and testing.
Supplementary data
To read the full content of this article, please download the PDF.
Moving image 1.
