Abstract
Aims: The SPIRIT II study randomised 300 patients in a ratio of 3:1 to receive either a XIENCE V or a TAXUS stent. The six month clinical and angiographic results have previously been reported. This paper presents the clinical follow-up of these patients to one year.
Methods and results: As a continuation in the assessment of the safety and performance of the XIENCE V Everolimus-Eluting Coronary Stent System (EECSS) enrolled patients were requested to return for clinical follow-up one year following the procedure to assess the occurrence of ischaemia driven major adverse cardiac events (MACE).
Of the 300 patients recruited at 28 sites in Europe, New Zealand and India, 223 were randomised to receive an EECSS stent and 77 a TAXUS paclitaxel eluting coronary stent system (PECSS). One-year clinical follow-up was obtained in 220 of the 223 patients in the EECSS group (98.7%) with one withdrawal prior to 180 days and two non-cardiac deaths (pulmonary malignancy and pneumonia) between six and 12 months, and in 76 of 77 (98.7%) PECSS group of patients (one patient had a missed 270-day and 1-year visit). Between six and 12 months there were no new occurrences of late stent thrombotic events in either group. There were no additional MACE events in the EECSS compared to, two, both ischaemia driven target lesion revascularisations (TLR) in the PECSS group.
Conclusions: The clinical safety of the XIENCE V EECSS stent observed at six months was sustained at one year. There were no additional thrombotic or MACE events in the EECSS group. Although not a primary endpoint, there was a significant difference in MACE favouring the EECSS compared to PECSS (2.7% versus 9.2%, P=0.04).
Introduction
Everolimus is an anti-proliferative agent that inhibits growth factor-stimulated cell proliferation by causing cell cycle arrest in the late G1 stage in the cell cycle.1 It is used as immunosuppressive therapy following heart and other solid organ transplantation, and has been shown to retard cardiac allograft vasculopathy.2
The feasibility of using everolimus (Certican®, Novartis Corporation, Switzerland) on a drug-eluting stent was demonstrated in the FUTURE-I3,4 and FUTURE II5 studies and more recently in the SPIRIT FIRST6 study which demonstrated both clinical safety and efficacy. In the SPIRIT FIRST study the angiographic in-stent late loss observed was 0.10 mm, a reduction of 88% relative to the bare metal stent at six months and an in-stent late loss of 0.24 mm at 12 months, a reduction of 71%.6,7
The SPIRIT II trial continued the assessment of the safety and performance of the XIENCE V everolimus-eluting coronary stent system (EECSS) by comparing it in a randomised fashion with the TAXUS paclitaxel eluting coronary stent system (PECSS) in the treatment of patients with a maximum of two de novo native artery lesions. At six months the rate of major adverse cardiac events (MACE) and angiographic late loss in the EECSS group was favorable, when compared with the PECSS group.8
This study evaluates the one-year clinical outcomes of patients enrolled in the SPIRIT II study treated with either an EECSS or a PECSS.
Methods
Patient population
Three hundred patients were randomised in a ratio of 3:1 to receive an EECSS (n=223) or a PECSS (n=77) at 28 sites in Europe, New Zealand and India. Included were patients 18 years of age and over, with evidence of myocardial ischaemia and a maximum of two de novo native coronary artery lesions, located in different major epicardial vessels. The target lesion(s) reference vessel diameter was required to be between 2.5 mm and 4.25 mm by visual estimation, < 28 mm in length, have a visually estimated stenosis of between 50-99% of the luminal diameter, and a Thrombolysis In Myocardial Infarction (TIMI) flow grade of 1 or more. Excluded were patients with a documented acute myocardial infarction within three days of the baseline procedure, a left ventricular ejection fraction of less than 30%, awaiting heart transplantation, or those with a known hypersensitivity or contraindication to aspirin, heparin, bivalirudin, clopidogrel or ticlopidine, cobalt, chromium, nickel, tungsten, everolimus, paclitaxel, acrylic and fluoropolymers. Patients with lesions that were aorto-ostial or left main stem, located within 2 mm of the origin of the left anterior descending- or left circumflex, heavily calcified, or had associated visible thrombus, were also excluded.
Study stent and procedure
The XIENCE V everolimus-eluting coronary stent system (manufactured by Abbott Vascular, CA, USA) is comprised of the ACS MULTI-LINK VISION coronary stent and delivery system, a thin fluoropolymer coating and everolimus. The ACS MULTI-LINK VISION stent (laser cut) consists of serpentine rings connected by links fabricated from a single piece of medical grade L-605 cobalt chromium alloy. Everolimus is blended with a fluoropolymer matrix that has been approved for use in blood contacting applications. The matrix of everolimus fluoropolymer contains 100 µg of everolimus per square centimetre of stent surface area. This system does not have a topcoat layer. The matrix is designed to release approximately 80% of the drug within 30 days after implantation in vivo.
For the SPIRIT II study, EECSS stents were available in lengths of 8, 18 and 28 mm, and diameters of 2.5, 3.0, 3.5 and 4.0 mm. Lesion lengths greater than 22 or < 28 mm were required to be covered by 2 stents; two 18 mm stents, or a 28 mm and an 8 mm stent. PECSS stents were available in lengths of 8, 12, 16, 20, 24, 28 and 32 mm and were of similar diameters to EECSS stents (2.75 mm were also available for PECSS).
Once preliminary angiography was performed and suitable lesion(s) identified, patients were randomised to receive a EECSS or PECSS stent. Due to packaging differences, physicians were not blinded to the device. Lesions were treated using standard interventional techniques with mandatory pre-dilatation and stent implantation at a pressure not exceeding the rated burst pressure. Post-dilatation was left to the discretion of the physician; however, if performed, was to be with balloons shorter than the stent length. In the event of a bailout procedure and requirement for an additional stent, this was required to be of the same type as the first implanted stent. Intravascular ultrasound (IVUS) was performed in a subset of 152 consecutive patients enrolled in pre-selected centres, after angiographically optimal stent placement had been achieved, and was repeated if additional post-dilatation was performed to optimise stent apposition and/or deployment. Periprocedural pharmaceutical treatment was administrated according to standard hospital practice. Procedural anticoagulation was achieved with unfractionated heparin or bivalirudin. The use of GPIIb/IIIa inhibitors was left to the discretion of the physician.
All patients enrolled into the study were maintained on 75 mg of clopidogrel daily for a minimum of six months and > 75 mg of aspirin daily for a minimum of one year following the procedure.
Follow-up
Patients were required to be reviewed at 30, 180, 270 days and one year following the procedure with further evaluations planned at two years and out to five years by protocol amendment. At outpatient visits, patients were questioned about the development of angina or the occurrence of any adverse events. Angiographic follow-up for all patients and IVUS in a subset of 152 consecutive patients (in selected centres) was performed at 180 days with the intention of repeating these investigations at two years in that subset of 152 patients. This report focuses on the one year clinical results.
Study endpoints
The primary endpoint of the study was angiographic in-stent late loss at 180 days. The secondary analyses included comparisons between the two groups, for measures including acute angiographic success, in-segment late loss, proximal and distal late loss, in-stent percentage volume obstruction, in-stent and in-segment percentage diameter stenosis and angiographic binary restenosis at 180 days and two years. Secondary clinical endpoints included ischaemia-driven MACE (cardiac death, myocardial infarction and ischaemia-driven target lesion revascularisation [ID-TLR]) either by CABG or PCI, evaluated at 30, 180 and 270 days, one and two year(s) after the index procedure. An amendment to the protocol will extend clinical follow-up to five years. These clinical endpoints were adjudicated by an independent clinical events committee (CEC) (Appendix I). In addition, a data and safety monitoring board (DSMB), not affiliated with the study, reviewed the data to identify any safety issues related to the conduct of the trial (Appendix I).
Definitions
All deaths that could not be clearly attributed to another cause were considered cardiac deaths.
A non-Q-wave myocardial infarction was defined as a typical rise and fall of CK-MB with at least one of the following: ischaemic symptoms, electrocardiographic (ECG) changes indicative of ischaemia (ST segment elevation or depression) or associated with a coronary artery intervention (if non-procedural/spontaneous MI, CK-MB > 2 times upper limit of normal; if post PCI, CK-MB ≥3 times upper limit of normal; if post CABG, CK-MB > 5 times upper limit of normal).
Ischaemia-driven target lesion revascularisation (ID-TLR) was defined as a revascularisation at the target lesion associated with any of the following: non-invasive positive functional ischaemia study (e.g. exercise testing or equivalent tests) or invasive positive functional ischaemia study (e.g. fractional flow reserve or coronary flow reserve); ischaemic symptoms and an angiographic percentage diameter stenosis (%DS) > 50% by on-line quantitative coronary angiography (QCA); or%DS > 70% by on-line QCA without ischaemic symptoms or a positive functional study.
Stent thrombosis, according to the study protocol, was categorised as acute (< 1 day), subacute (>1 day < 30 days) and late (>30 days). It was defined as a clinical presentation of an acute coronary syndrome with angiographic documentation of complete occlusion of a previously successfully treated target vessel and/or angiographic documentation of a flow limiting thrombus within or adjacent to a previously successfully treated target lesion. In the absence of angiography, cardiac death, acute myocardial infarction (AMI) in the territory of the stented vessel(s), or an AMI that could not be clearly attributed to a non-target vessel, were included in the composite analysis for stent thrombosis.
Subsequent to writing of the study protocol a new definition of stent thrombosis, generated by the Academic Research Consortium (ARC), has become accepted by the interventional community.9 Thereby stent thrombosis is defined as angiographic confirmation of the presence of thrombus originating in the stent or in the segment 5 mm proximal or distal to the stent and presence of at least one of the following criteria within a 48 hours time window: 1) acute onset of ischaemic symptoms at rest; 2) new ischaemic ECG changes suggestive of acute ischaemia; 3) a typical rise and fall in cardiac biomarkers; 4) non-occlusive thrombus; or 5) occlusive thrombus.
All stent thrombotic events occurring up to one year follow-up were re-adjudicated by the CEC, and stent thromboses defined both as per study protocol and by the ARC definition are reported in the results section below.
Statistical methods
The rationale for sample size calculations for this study has previously been reported.8 In this paper, binary variables are presented as percentages and compared using the Fisher’s exact test. Continuous variables are presented as mean±standard deviation and compared using the Wilcoxon two-sample test. The hypothesis testing for the primary endpoint was performed using a one-sided non-inferiority test with asymptotic test statistic based on a non-inferiority margin of 0.16 mm. As non-inferiority was shown, superiority was also tested with a two-sided t-test, as planned in the protocol. Primary analysis was based on the analysis lesion (target lesion for single lesion treatment and one randomly selected lesion for dual lesions treatment).
Results
Patient population and lesion characteristics
The baseline demographic, clinical and angiographic characteristics of the treatment groups have previously been reported8 and are summarised in Table 1.
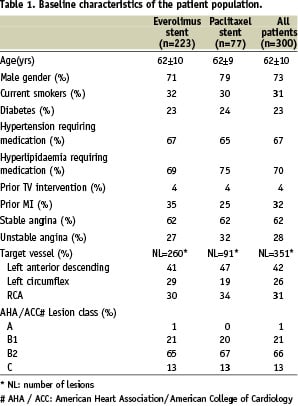
There were no significant differences between treatment groups in any of the tabulated characteristics. A flowchart of the clinical follow-up through one year, on an intention to treat basis (223 patients EECSS group, 77 patients PECCSS group), is displayed in Figure 1.
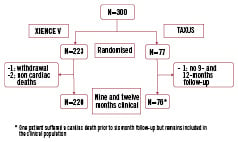
Figure 1. Flowchart of patient follow-up.
One patient crossed over from the EECSS to the PECSS arm but is analysed in the EECSS arm (Intention to Treat)
One-year clinical follow-up was obtained in 220 of the 223 EECSS group patients (98.7%) with one withdrawal prior to 180 days and 2 non-cardiac deaths (pulmonary malignancy and pneumonia) between six and 12 months. In the PECSS group 76 of 77 (98.7%) recruited patients completed one-year follow-up with one patient who did not attend nine and 12-month follow-up visits.
Six-month clinical and angiographic outcomes
The six-month outcomes have previously been reported8 but are summarised below. Hierarchically, for the intent-to-treat population, in the EECSS arm, two (0.9%) non-fatal non-Q wave MIs and four (1.8%) ID-TLRs by PCI were identified compared to one cardiac death (1.3%), two (2.6%) non-fatal non-Q wave MIs and two (2.6%) ID-TLRs by PCI in the PECSS arm. The total hierarchical MACE rate was 2.7% (6/222) in the EECSS eluting arm vs. 6.5% (5/77) in the PECSS arm. In addition there were two (0.9%) ID-TVRs (non-target lesions) in the everolimus arm and none in the paclitaxel arm. There were no occurrences of acute or subacute stent thromboses in either arm. One case of late stent thrombosis occurred in the EECSS arm at 53 days and one in the PECSS arm at 56 days post-stent implantation resulting in the latter patients death. Both patients were taking dual antiplatelet therapy at the time of their thrombotic event.
At six month follow-up the in-stent late loss for the analysis lesions was 0.11±0.27 mm for the EECSS arm and 0.36±0.39 mm for the PECSS (non-inferiority p<0.0001, superiority p<0.0001). For all lesions the in-stent late loss was 0.12 mm versus 0.37 mm for the EECSS and the PECSS groups respectively. In the EECSS group the in-stent diameter stenosis was 16%, in-stent binary restenosis 1.3%, neo-intimal volume 3.8 mm and volume obstruction 2.5%. In the PECSS group the in-stent diameter stenosis was 21%, the in-stent binary restenosis 3.5%, neo-intimal volume 14.4 mm and volume obstruction was 7.4%.
One-year clinical outcomes
Between six months and one year no further hierarchical MACE events occurred in the EECSS group and two in the PECSS group – both ischaemia driven target lesion revascularisations by PCI. The hierarchical analysis of the one-year clinical results showed that the total MACE rates for the procedure through the one year interval were 2.7% in the EECSS group and 9.2% in the PECSS group (p=0.04). These results are displayed in Table 2.
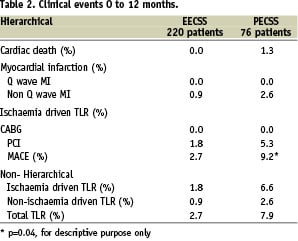
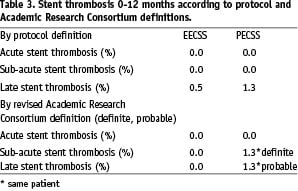
A total of two cases of late stent thrombosis, as defined by the study protocol, were reported through to 180 days post-procedure, of which one was in the EECSS group and one in the PECSS group. No new cases of late stent thromboses were observed in either group between 180 days and the one-year follow-up time point.
When the new definition of stent thrombosis, according to the ARC was applied,9 the one patient in the EECSS with a stent thrombosis according to the protocol, was no longer defined as having such. This patient suffered an acute coronary syndrome following the index procedure with later angiographic documentation of complete occlusion of the target lesion, but documentation of the occlusion occurred > 48 hours (53 days later) after the acute coronary syndrome, thus not fulfilling the definition.
In the PECSS group, one patient was regarded not to have suffered a sub-acute stent thrombosis according to the study protocol definition but was defined as having a definite stent thrombosis according to the ARC definition. This patient suffered a non-Q MI and within 48 hours underwent angiography, which showed TIMI grade 3 flow and thrombus within the stent. This patient suffered also a late stent thrombosis according to both the study protocol definition and the ARC definition. These findings are summarised in Table 3.
Discussion
The clinical efficacy and safety of an everolimus-eluting stent has been strengthened by the SPIRIT II study which confirms the earlier work on the FUTURE I and II3-5 and SPIRIT FIRST studies.6 This study follows the natural progression of the rigorous evaluation of a next generation drug eluting stent by comparing the XIENCE V stent in a randomised way with the TAXUS stent, which has, along with the CYPHER sirolimus eluting stent become universally accepted into routine clinical practice.10
The six month clinical, angiographic and IVUS results showed the first glimpse at the clinical impact for a next generation drug eluting stent. This non-inferiority randomised trial not only met its primary endpoint, but also demonstrated the superiority of the EECSS over the PECSS in terms of in-stent late loss. In addition, the IVUS results showed that the XIENCE V stent was more effective at reducing neointimal hyperplasia than the TAXUS stent. Additionally there was a very low rate of hierarchical MACE events – 2.7% in the EECSS group and 6.5% in the PECSS group.8 The one year clinical follow-up has shown that these excellent outcomes have been maintained with no change in the MACE rate for the EECSS patients, remaining at 2.7% and increasing from 6.5% to 9.2% in the PECSS group of patients, consisting of two ID-TLR treated by PCI.
The results of the SPIRT III study, undertaken in the United States and Japan, have recently been presented.11 In this study, patients were randomised in a ratio of 2:1 to receive a XIENCE V stent or a TAXUS PECSS stent. At eight month angiographic follow-up, the late loss of 0.14 mm was significantly less in the EECSS group when compared to the PECSS patients in whom the late loss was 0.28 mm. This not only met the criterion for non-inferiority but was also statistically superior. There were also significantly fewer MACE at nine months in the EECSS group compared to the PECSS patients (4.6% versus 8.1%, p=0.028).11
As has been seen in other studies such as RAVEL,12 the results of later studies are less striking as the patient populations enrolled converge from those with favourable “research” lesions to “real world” lesions as investigators become less cautious about the new technology. Despite this the SPIRIT III results show a clear clinical advantage of the XIENCE V stent over the TAXUS drug eluting stent.
These results provide a great deal of optimism that the XIENCE V stent is at least as good as the currently available drug eluting stents. We must remain cognizant of the fact that the established commercially available stents have a much longer research history extending for periods of five years and beyond.13 The clinical question then arises: at which point can we feel comfortable and confident in the safety and efficacy of a new stent with enough evidence base for it to enter routine clinical practice? In the case of the XIENCE V stent system, the drug everolimus and the VISION stent platform are both well established.
Often it takes more than non-inferiority to alter practice, in that clinicians require evidence of superiority before changing from the familiarity and comfort of established practice. To date the safety and efficacy of the XIENCE V stent has not been doubted and is reinforced by these new results. Patients enrolled in the SPIRIT II trial will soon begin returning for two year clinical and angiographic follow-up, which will significantly add to the XIENCE V database of evidence.
Further SPIRIT studies of the EECSS are in progress. In the U.S., the SPIRIT IV study, a modified extension of SPIRIT III, intends to enrol 3,690 patients to provide additional data on safety and efficacy in patients with up to three de novo native coronary artery lesions. Again patients will be randomised to receive an EECSS or PECSS to obtain data on patients with more complex coronary anatomy, bringing the research population closer to “real world” situations. The SPIRIT V international evaluation plans enrolment of 3,000 patients and consists of both a randomised study of diabetic patients as well as a registry of use of the EECSS in day-to-day clinical settings. SPIRIT WOMEN is the world’s first drug eluting stent study to include only women. This will evaluate patient and disease characteristics specific to women and study clinical outcomes following stenting with the EECSS.
The recent concerns over increased rates of stent thrombosis prompted us to carefully evaluate and adjudicate the thrombosis rates in the SPIRIT II study. Stent thrombosis as defined by the study protocol, which predated the ARC definitions, as well as the thrombosis rates according to the new definition have been reported. The study numbers are too small to comment on rates of thrombosis and to compare the rates between stents but the trend of reduced rates in the EECSS group is certainly encouraging. There were no additional late stent thrombosis events between six months and one year. Again we look forward to the results of the two-year follow-up, and even further with the recently extended amendment to follow SPIRIT II patients for a period of five years.
Conclusions
The clinical safety of the XIENCE V stent observed at six months was sustained at one year. This was demonstrated by no new observations of either late stent thrombotic events or MACE in the EECSS group. Although not the primary endpoint, there was a significant benefit in MACE favouring the XIENCE V EECSS over the TAXUS PECSS.
Acknowledgements
Sponsor: Abbott Cardiovascular Systems, an Abbott Vascular Company, Santa Clara, California, USA.
Principal Investigator: Patrick W. Serruys (The Netherlands).
Steering Committee - Patrick W. Serruys (Principal Investigator and Chairman, Rotterdam, The Netherlands); Gary Johnson (Vice President of Regulatory Affairs, Pre-Clinical Research, Clinical Research and Reimbursement, Cardiac Therapies, Abbott Vascular); Marcus Wiemer, Bad Oeynhausen, Germany; Eulogio Garcia, Madrid, Spain; John Ormiston, Auckland, New Zealand.
Data Safety Monitoring Board (DSMB) - Jan G.P. Tijssen, Amsterdam, The Netherlands; Thierry Lefèvre, Massy, France; Philip Urban, Meyrin-Geneva, Switzerland.
Clinical Events Committee (CEC) - Claude Hanet, Clinique Universitaire de Saint-Luc, Brussels, Belgium; Dougal McClean, Christchurch, New Zealand, Victor Umans, Medical Center Alkmaar, The Netherlands
Angiographic and IVUS Core Laboratories: Cardialysis BV, Rotterdam, The Netherlands.
Data Coordination Centre and Site Monitoring: Abbott Cardiovascular Systems, an Abbott Vascular Company, Diegem, Belgium.
The following investigators and institutions participated in the SPIRIT II Trial:
J.J. Piek, Academisch Medisch Centrum, Amsterdam, The Netherlands; P. Ruygrok, Green Lane Cardiovascular Service and Mercy Hospital, Auckland, New Zealand; J. Neuzner, Klinikum Kassel, Kassel, Germany; A. Seth, Max Devki Devi Heart & Vascular Institute, New Delhi, India; J. Schofer, Universitäres Herz-und Gefässzentrum Hamburg, Hamburg, Germany; M. Wiemer, Herzzentrum Bad Oeynhausen, Bad Oeynhausen, Germany; G. Richardt, Segeberger Kliniken GmbH, Bad Segeberg, Germany; D. Carrié, Hôpital de Rangeuil CHU, Toulouse, France; L.Thuesen, Skejby Sygehus, Aarhus, Denmark; E. Camenzind, R.V. Hôpital Cantonal Universitaire de Geneve, Geneva, Switzerland; H. Kelbaek, Rigshospitalet, Copenhagen, Denmark; P.W. Serruys, Erasmus Medical Center, Rotterdam, The Netherlands; C. Macaya, Hospital Clinico San Carlos, Madrid, Spain; J. Berland, Clinique Saint Hilaire, Rouen, France; M. Desaga, Amper Kliniken AG-Klinikum Dachau, Dachau, Germany; F. Van den Branden, A.Z. Middelheim, Antwerpen, Belgium; K. Rasmussen, Aalborg Sygehus Syd, Aalborg, Denmark; H. Suryapranata, Isala Kliniken Locatie Weezenlanden, Zwolle, The Netherlands; V. Legrand, C.H.U. de Liège Sart Tilman, Liège, Belgium; W. Ruzyllo, National Institute of Cardiology in Warsaw, Warsaw, Poland; A. Manari, Azienda Ospedaliera Santa Maria Nuova, Reggio Emilia, Italy; C. Spaulding, Hôpital Cochin, Paris, France; M. Suttorp, St. Antonius Ziekenhuis Nieuwegein, Nieuwegein, The Netherlands; J. Boland, C.H.R. La Citadelle, Liège, Belgium; K. Huber, Wilhelminenspital der Stadt Wien, Vienna, Austria; E. Garcia, University Hospital Gregorio Maranon, Madrid, Spain; J.A.M. te Riele, Amphia Hospital, Breda, The Netherlands.
