Abstract
Aims: The Custom NX® 36 Drug Eluting Stent (DES) System is designed to treat coronary lesions via in situ stent length customisation. The stent evaluated in this study consists of nine interdigitated stent segments, each 4 mm in length, coated with a biodegradable formulation of Biolimus A9® and Poly-Lactic-Acid, a biodegradable drug carrier, and loaded into a unique sheath protected, integrated balloon delivery system.
Methods and results: The objective of this non-randomised prospective multicentre CUSTOM I Trial was to demonstrate the safety of in situ stent length customisation in 30 consecutive patients. Angiographic (QCA) and intravascular ultrasound (IVUS) follow-up was performed at two cohort time intervals: four (n=10) or eight (n=20) months. Mean lesion length and reference vessel diameters were 17.7±9.6 mm and 2.6±0.3 mm, respectively. Procedural success was 93%. There were three MACEs in the study population from enrollment through to twenty-four month follow-up. The in-hospital MACE rate was 2/30, with non-Q-wave myocardial infarctions events in two patients who recovered without further sequelae. At five months, one patient who had crossed over initially to PTCA required CABG surgery. Results from QCA and IVUS assessments at four and eight months showed no binary restenosis, mean in-stent late luminal loss of 0.25±0.23 mm and 0.26±0.23 mm.
Conclusions: This first evaluation of the new customisable Biolimus A9-eluting Custom NX stent suggests safety and efficacy through twenty-four month follow-up. Further evaluations are warranted to confirm the overall favourable outcomes.
Introduction
First generation drug eluting stents (DES) have demonstrated that they can significantly reduce neointimal hyperplasia formation when compared to bare metal stents, resulting in improved clinical outcomes with lower target vessel or lesion revascularisation rates1,2. The Custom NX® 36 DES System (XTENT® Inc., Menlo Park, CA, USA) belongs to a new generation of stent catheter systems that has been specifically developed to address limitations of existing DES systems. This device combines a unique delivery and integrated balloon system with a drug eluting stent matrix. The stent features a coating formulation specifically designed for endovascular use, which consists of a sirolimus derivative, Biolimus A9® that binds to the cytosolic immunophilin FK506-binding protein 12 (FKBP12) and inhibits growth factor driven smooth muscle and T-cell proliferation, coupled with a biodegradable D, L-Poly Lactic Acid (PLA) polymer licensed from Biosensors International, Singapore3. Biolimus A9 offers enhanced lipophilicity, enabling increased uptake in local target tissues and reduced presence in the systemic circulation.
Prior to inception of this study; preclinical feasibility, proof of concept, biocompatibility, safety, pharmacokinetics and toxicity studies were undertaken by Xtent and independent investigators4,5. The results demonstrated that balloon-expandable, modular-segmented, cobalt alloy stents coated with the biodegradable polymer PLA and the rapamycin derivative Biolimus-A9 produced consistent and reproducible inhibition of neointimal formation in the porcine coronary model4. Long-term biocompatibility was established by low injury and inflammation scores as well as safety as seen in high animal survival rates through 270-days. These studies represented an assessment of more than 250 stent implants in 110 animals.
Biosensors International initiated a first-in-man trial STEALTH I in 2003-2004 to evaluate the safety and efficacy of the Biomatrix stent coated with the same formulation as used for the CUSTOM NX, in the treatment of single de novo lesions in native coronary arteries. The study results established a low restenosis rate (3.9% vs. 7.7%) and a significant decreased late loss (0.26 mm vs. 0.74 mm) in Biolimus A9 DES patients when compared with the control6. Significant reductions were also observed in percentage diameter stenosis at follow-up (23.5% vs. 30.9%) and % neointimal volume obstruction determined by intravascular ultrasound (IVUS) (2.6% vs. 23.5%). These results demonstrate the clinical safety and efficacy of Biolimus A9 and a PLA polymer formulation at six months. The Xtent formulation is provided by Biosensors International and has been used for vascular stent applications by Biosensors (Biomatrix® stent) or other licensees (Axxess™ Plus Stent, Devax Inc., Lake Forest, CA, USA and Noburi™ stent, Terumo Corp., Tokyo, Japan).
The aim of this first-in-man trial was to evaluate safety and efficacy of the Custom NX® DES system to treat patients with coronary lesions via in situ stent length customisation.
Methods
Device description
The Custom NX® DES System allows for customised implantation of balloon expandable cobalt chromium DES stents (Figure 1).
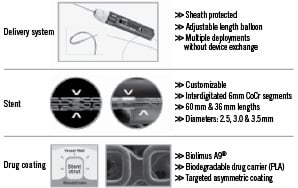
Figure 1. Custom NX® DES system
The device enables the operator to adjust the stent length to the intended lesion length within the coronary vasculature. This determination occurs in situ, in the site of intended stent implantation. A handle at the proximal end of the catheter and external to the patient, can be manually operated for stent exposure, separation and deployment. At the distal end of the device protected by a sheath there is 36 mm of stent in nine 4 mm incremental segments premounted on a semicompliant balloon. Each cobalt chromium stent segment is coated with a drug product formulation consisting of Biolimus A9 and the PLA biodegradable drug carrier. In order to customise the stent length, a switch located on the handle is manipulated to activate segment separation as determined in situ. By connecting the handle end to a standard angioplasty indeflator, the interventionalist can effectively deploy the integrated balloon at desired pressures within recommended product compliance ranges. Additional handle adjustment allows the operator to shorten the balloon length to permit higher pressure post-dilatation within the stented segment.
Trial design
CUSTOM I was a nonrandomised, prospective multicentre first-in-man study that enrolled 30 patients with symptomatic ischaemic heart disease characterised by discrete de novo coronary artery lesions (up to two lesions) with reference vessel diameters ranging between 2.6-3.1 mm. Thirty patients were enrolled at three European investigational sites from June 10th, 2005 to July 13th, 2005. The primary objective of this study was to gather data regarding the clinical performance of the device in patients treated for coronary artery disease with in situ customisation of stent length.
The primary study endpoint was the rate of major adverse cardiac event (MACE) at eight months, defined as: cardiac related death, any myocardial infarction, and clinically driven target lesion revascularisation. The secondary safety endpoints included MACE rates at 30 days and four months, the incidence of bleeding and/or vascular complications, and the incidence of stent thrombosis at 30 days and eight months. The quantitative coronary angiographic (QCA) endpoints at four or eight months included binary restenosis, late loss, loss index, and minimal luminal diameter (MLD). Major exclusion criteria included: ejection fraction < 30%, myocardial infarction within 72 hours, previous stenting within target vessel, ostial, left main or bifurcation lesions, and target lesion involving a side branch > 2.0 mm in diameter.
As shown in Figure 2, all patients were scheduled to undergo clinical follow-up at one, four and eight months with annual follow-up through five years post index procedure.
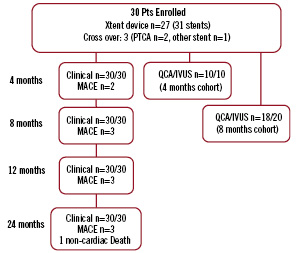
Figure 2. CUSTOM I study flow chart
The first ten patients enrolled in the study were scheduled to undergo angiographic follow-up at four months post procedure. The remaining patients (n=20) were scheduled to undergo angiographic follow-up at eight months post procedure.
The study was conducted according to the principles of the Declaration of Helsinki and the European Union recognised standard ISO-14155: 2003. A signed informed consent was requested and obtained from each patient prior to inclusion in the study.
Procedural steps
Patients received aspirin (100-500 mg) and clopidogrel (300 mg) prior to percutaneous coronary intervention and were continued on aspirin (100 mg) and clopidogrel (75 mg) as dual antiplatelet regime for a recommended minimum of 90 days. Intravenous heparin (5,000-10,000 units) were administered during the procedure to maintain an activated clotting time (ACT) >250 sec. Preprocedure angiograms were recorded per angiographic core lab work instruction. Direct stenting was not permitted per protocol. Target lesions were predilated with a commercially available balloon system. The Custom NX DES catheter system features allowed for up to 36 mm of stent length, and is compatible with a 7 Fr guide catheter and 2.6 mm-3.1 mm estimated reference vessel diameter. In order to assess stent expansion and apposition, the protocol required a post procedure intravascular ultrasound (IVUS). ECGs were taken immediately post procedure and pre-discharge, as were cardiac enzymes at six and 24 hours post procedure.
Definitions and statistical analysis
Categorical variables were to be summarised using counts and percentages. Continuous variables were to be summarised using mean, standard deviation, minimum value, maximum value, and median. All safety data were to be summarised as part of the intent-to-treat analysis.
Data management
Data monitoring was performed by an independent clinical research organisation (Krauth Medical, Hamburg, Germany). Angiograms were analysed by an independent angiographic core laboratory at the University of Florida (Jacksonville, FL, USA). Intravascular ultrasound (IVUS) data was analysed by Stanford University IVUS core laboratory (Palo Alto, CA, USA). Clinical events were adjudicated by an independent committee at Cardiovascular Research Foundation, New York City (New York,USA).
Results
Baseline and procedural results
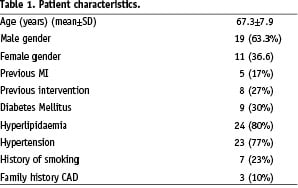
A total of 27 patients were exclusively treated with the Custom NX 36 DES System while three patients received another stent (n=1) or crossed over to percutaneous transluminal coronary angioplasty (PTCA) treatment (n=2). All patients underwent scheduled clinical follow-up as pre-determined in the study protocol. A total of 28 patients underwent angiographic follow-up as pre-determined in the study protocol. Two patients did not undergo angiographic follow-up due to patient refusal (n=1) or angiographic procedure prior to the follow-up time point (n=1). The demographic characteristics of the patients enrolled in the CUSTOM I trial are provided in Table 1.
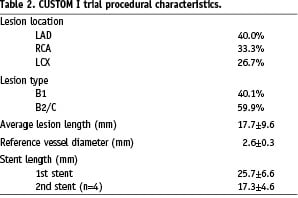
The procedural characteristics of the patients enrolled in the CUSTOM I trial are shown in Table 2.
Four patients enrolled in the study were treated with two Custom NX DES devices resulting in two customisable stent implantations in these patients.
Safety results
MACE events were reported in three patients. Two of those patients experienced a cardiac enzyme elevation post procedure resulting in a non-Q-wave myocardial infarction as defined by the protocol. Significant for the first patient was one transient episode of peri-procedural ST elevation, which was resolved with nitrates followed with initiating IV Integrillin therapy. The proximal LAD lesion was successfully treated with 32mm Custom NX stent. The presumed mechanism of this event is unknown. The second patient presented with multiple risk factors including previous CABG surgery, diabetes mellitus and EF of 35%. The lesion required three predilation attempts before the proximal circumflex lesion could be successfully implanted with 28 mm Custom NX stent. In both cases no post procedure ECG changes were observed. Both patients were discharged from the hospital free of symptoms and no other cardiac events have been reported through twenty four month follow-up. The third patient did not receive a Custom NX 36 DES System during the index procedure, rather balloon angioplasty following failed attempts to cross the target lesion with not only with the investigational device, a commercially available drug eluting and a bare metal stent as well. This patient later received coronary by-pass surgery five months post procedure for recurrence of angina symptoms and worsening of cardiovascular disease in non-target vessels. No new events have been reported for this patient through the two year follow-up. Lastly, between the first and second year follow-up period a non-cardiac death was reported in a patient with end stage bronchial carcinoma who was under chronic anticoagulation treatment and suffered an extensive subdural haemorrhage following a traumatic fall.
All events were reviewed by an independent clinical event committee (CEC) who adjudicated the nature of the event and its possible relationship to the procedure or to the device. MACE events reported were not adjudicated as device related. No stent thrombosis was reported at any of the study time points up to two years. Five other cardiovascular adverse events during the course of the follow-up were reported. One patient underwent a non-target vessel revascularisation 20 months post index procedure. One patient was hospitalised for cardiac decompensation and treated medically. Three patients were hospitalised for chest pain and underwent coronary angiography with no interventions indicated.
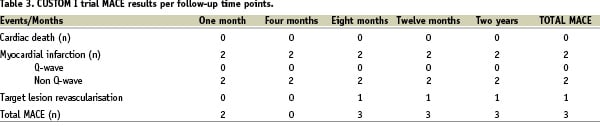
Efficacy and performance results
A total of 30 patients presenting 34 coronary artery lesions were enrolled in the CUSTOM I trial. The procedural success rate was 93% effectively achieving final diameter stenosis of <15% (visual estimate) with any device, and the occurrence of two MACE during index hospitalisation. Device success rate was 90% given three cases where the Custom NX 36 could not deliver a customisable stent. In one patient, the presence of a previously implanted stent in the target lesion was noted at the time of Custom NX 36 advancement into the vessel. This being an exclusion criterion, the investigator decided to stop the CUSTOM I procedure and cross-over to a marketed drug eluting stent. Because the treatment attempt included insertion of the Custom NX 36 device into the patient vasculature, the patient was de facto enrolled in the study on the “intent-to-treat” basis of the study protocol. The second patient presented with a narrow proximal RCA lesion that the Custom NX could not cross. QCA core lab RVD measurement suggests diameter sizing mismatch. The third patient’s circumflex lesion had a significant tortuous bend preventing lesion crossing with the Custom NX system, as well as other commercially available drug eluting and bare metal stent. Both of the later patients were subsequently treated with percutaneous balloon angioplasty. One of which remained free of symptoms through two year follow-up and the last patient underwent cardiac surgery for recurrence of symptoms five months post procedure and has been reviewed in the prior safety paragraph.
A total of 10 and 18 patients underwent angiographic follow-up, at four and eight months post procedure respectively, as pre-determined by the study protocol. The secondary angiographic endpoints used to confirm the efficacy of the Biolimus A9 PLA formulation were the incidence of binary restenosis, the measurement of late luminal loss, loss index and late absolute minimal luminal diameter at four or eight months via quantitative coronary angiographic (QCA). The results are summarised in Table 4.
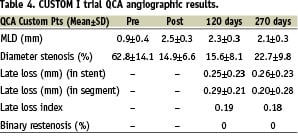
A vascular examination using intravascular ultrasound (IVUS) was also performed during the angiographic follow-up. Those results included quantitative and qualitative assessment of the in situ customisable stent in order to evaluate deployment characteristics and to measure the efficacy of the Biolimus A9 and PLA coating formulation. The measurements included the determination of the minimum lumen area within the body of the stent post implant and, the amount of obstruction present within the stent at follow-up. Baseline and follow up IVUS images were available for nine and 10 patients at the four and eight month follow-up periods respectively. The results of those observations are provided in Tables 4 and 5 below.

Discussion
The CUSTOM I trial objective was to demonstrate the safety and efficacy of the Custom NX® 36 DES system. One of the novel features of this device is the in situ customisation of the stent length. Current commercially available stents are all fixed in length, which present the following potential limitations: inaccurate placement of stents, overlapping of stents to cover long lesions, and anatomical distortion of the coronary artery due to stent rigidity. In contrast, while the Custom NX device is being positioned in the patient’s vascular system using fluoroscopic imaging, the physician can navigate complete coverage of the lesion and adjust as needed in situ. The ability to customise with the Custom NX device allows the physician to incrementally increase or decrease the desired stent length to be deployed while in the patient’s vascular system. The benefits of this feature can reduce the incidence of geographic miss and the resulting problems associated with restenosis and reintervention.
Moreover, the Custom NX stent’s proprietary design of multiple, small individual segments that are interdigitated, provides enhanced flexibility. This flexibility is important during delivery of the customisable stent as well as after stent deployment. The Custom NX stent is made from cobalt chromium and designed to provide the flexibility needed to better maintain the vessel’s natural contour as well as move and flex with contractions of the heart as compared to current stent technologies that exhibit higher rigidity. Another unique feature of this device is how the stent is delivered to the lesion within a sheath that protects the stent and its coating from damage as it is pushed through a patient’s vascular system. Current delivery systems leave stents exposed during placement that may result in outer coating damage during the navigation process through bends of the vascular system.
In addition, several of the currently available stents have durable polymer coatings which have been associated with delayed endothelialisation and hypersensitivity reactions. The Custom NX device utilises a biodegradable drug carrier and Biolimus A9 elution. The biodegradability of the BA9/PLA coating formulation allows for degradation of the drug and coating within the stent surrounding tissues. Ultimately the implant returns to a profile comparable to a bare metal implant for which known long term data and broad history has been reported. Due to the novel approach of the Custom NX system, careful clinical evaluation in the first-in-man setting has been performed to make sure that no adverse reactions could be associated to the Custom NX DES System or that the expected long term benefits of the stent implants were not impacted by the device manufacturing/processing or the operating principles leading to customisation.
CUSTOM I demonstrated both safety and efficacy of this technique in a first-in-man application.
Of the 30 patients enrolled in the study, two cardiac enzyme elevations were reported and in both cases, the patients recovered without reports of any additional adverse events through to the two year follow-up. This type of complication was not unanticipated and has been reported at similar rates in other studies involving drug eluting stents7-16. In some cases those studies involved stents with the same PLA polymer9,11,12. There was no device mechanical failure in the study that resulted in patient complications.
No patient treated with the Custom NX device experienced restenosis or revascularisation of the treated lesion. One death was reported between the one and two year follow-up that was adjudicated by an independent clinical events committee as being non-cardiac. There was no late thrombosis reported through two year follow-up post procedure. The angiographic observations were in line with those made in other studies completed with Biolimus A9/PLA platforms7-9,11,12.
Conclusion
The first-in-man study CUSTOM I suggests safety, efficacy and durability of implanting the unique customisable Biolimus A9-eluting Custom NX System in patients with de-novo coronary lesions. Further research is warranted to confirm these favourable clinical results.
