Introduction
Drug-eluting stents (DES) were introduced into clinical practice in 2002 in order to reduce restenosis that occurred in 15-25% of patients receiving bare-metal stents (BMS).1-3 Subsequent trials with different types of DES confirmed their efficacy in this regard.4 However, late stent thrombosis was reported as early as 2004, typically in patients discontinuing dual anti-platelet therapy.5 At the European and World Congress of Cardiology in Barcelona 2006, alarming data were presented on a worse long-term prognosis following DES implantation compared with BMS.6,7 As a result both randomized controlled trials and registry data were scrutinized to validate these concerns, bearing in mind the differential values of both types of studies.8,9 Furthermore, the worldwide discussion on the long-term safety and efficacy of DES triggered the European Society of Cardiology together with the European Association for Percutaneous Cardiovascular Interventions to organize a forum on DES. On 27 and 28 September 2007, key opinion leaders in (interventional) cardiology and representatives from industry and regulatory bodies gathered in the European Heart House with the intention to review: (i) the most recent data on the long-term efficacy (reduction of restenosis, re-intervention) and safety (late stent thrombosis, myocardial infarction, mortality) of DES and its effects on outcome (survival, event-free survival), (ii) specific indications for DES; (iii) health economical analyses currently performed with DES; (iv) the DES registration process in Europe; (v) current and possible future trial designs. The overall goal was to provide general recommendations to the medical community for the use, clinical development, and future assessment of DES.
Safety
In several randomized controlled trials comparing sirolimus-eluting stents (SES) or paclitaxel-eluting stents (PES) and BMS, increased rates of death or myocafigrdial infarction were observed at follow-up, beyond the first year,6-8,10 while no excess of such events occurred in the first year.11-14 Subsequently, several pooled analyses of individual patient-level data from these and other trials indicated similar safety up to 4 years.15-18
In particular, the long-term safety of DES was assessed in a network meta-analysis, including 38 randomized controlled trials in over 18,000 patients. While there was no difference in mortality up to 4 years in patients initially treated with either BMS, SES, or PES, the SES was associated with a 19% lower risk for myocardial infarction (MI) as compared with BMS and 17% as compared with PES.
In the latest report of the Swedish SCAAR registry (n=35 262) presented at the ESC congress in Vienna 2007, the incidence of death and MI were similar between DES and BMS up to 4 years.19 However, the survival curves showed a short-term survival benefit with DES up to ~1 year followed by a late catch-up. Thus, the apparent early benefit is lost by an excess of subsequent events. A pattern of lower initial and increased late mortality rates following DES use was reported by RAVEL2,7 and the Basel Stent Kosten Effektivitäts Trial (BASKET-LATE; DES: n=281) investigators.10 While there was no significant difference in death and MI at 3 years, this was the net result of a higher 6 month survival and a higher incidence of late (>6 months) death, or MI following DES. According to the authors, this latter might be the consequence
of the discontinuation of clopidogrel at 6 months.10 It is noteworthy that the impaired outcome with DES that was reported earlier by the SCAAR investigators in patients treated up to 2004 was no longer present in the current analysis, which included patients treated in 2005.9,19 Additionally, the Western-Denmark registry (n=12,395), Ontario registry data (n=13,353), and data from the state of Massachusetts demonstrated a trend towards a lower mortality rate following DES in the first months, which was maintained up to 2-3 years.20-22 Similarly, in an analysis of 6,129 consecutive patients from Rotterdam, the Netherlands, the use of SES, but not PES, was associated with a significantly higher 3 year survival as compared with BMS.23 The survival benefit with SES became apparent as early as 3 months. The ENDEAVOR programme, evaluating a new zotarolimus-eluting stent, showed significantly lower cardiac death and MI rates for the DES as compared with BMS up to 3 years.24
It should be appreciated that follow-up in different trials, meta-analyses, and registries varies from 1 or 2 years to 5 years. These differences should be taken into consideration when comparing these studies. Furthermore, it should be appreciated that there may be important differences in both efficacy and safety among different types of DES as well as among BMS (Figure 1). Not all DES are equal, nor are BMS! If the trend of an early benefit with some DES, with a later excess of death and MI as observed in some studies, would persist, long-term outcome beyond 5 years might gradually favour some types of BMS. This is, of course, highly speculative! Nevertheless, it is desirable that the investigators will continue
to provide such very long-term follow-up data.
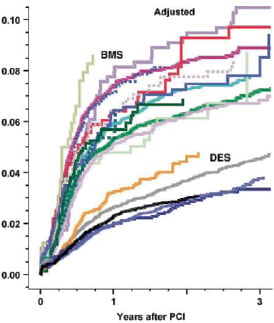
Figure 1. Restenosis at clinically driven re-angiography (data from the SCAAR registry presented by Dr S. James; ACC 2008). There are major differences for in-stent restenosis in BMS ranging from 7% to 10.5% at 3 years (upper series of curves) as well as among DES, ranging from 3.3% to >4.6% at 3 years (lower 5 curves).
Late stent thrombosis
Stent thrombosis has been linked to a wide variety of pathophysio-logical mechanisms and clinical and procedural risk factors.6,25-28 It has been associated with mortality rates varying between 15 and 45%.29,30
Data from registries and meta-analyses6-10,16-24 indicated that there is no difference in the risk of early (<30 days) and late (>30, <365 days) stent thrombosis between DES and BMS, but that an excess risk emerges after more than 1 year of follow-up (very late stent thrombosis). The incidence of angiographically documented stent thrombosis in the combined Bern-Rotterdam experience with PES and SES was 0.6% per year, without any sign of reduction up to 4 years after stent implantation, and with slightly higher rates for PES than SES.31 A comparable rate of 0.5% was observed in the SCAAR registry.9 This suggests that endothelial healing remains impaired up to 4 years, at least in some patients and/or with some DES. Of interest were the recently presented 2 and 3 year follow-up of the ENDEAVOR I, II, and III trials, which showed remarkably low rates of stent thrombosis (0.0-0.3%) and no cases of stent thrombosis after 30 days.24 Of note, these pivotal studies evaluating the relative safety and efficacy of the zotarolimus-eluting stent were restricted to relatively simple lesions and patients. It is possible that this zotarolimus-eluting stent has a better safety profile than SES
or PES. However, no direct comparative data are available. The results of the large E-Five registry and the 8,800 patients randomized PROTECT trial are eagerly awaited.
It should be appreciated that documentation of stent thrombosis by angiography underestimates the real incidence of such event, since some of the patients with stent thrombosis develop MI, or die without angiographic documentation of stent thrombosis. To address this issue, new definitions were formulated by a consortium of interventional cardiologists from both sides of the Atlantic, representatives of the Food and Drug Administration (FDA), clinical research organizations, and representatives from major stent manufacturers (Figure 2).32

Figure 2. ARC definitions of stent thrombosis.
Efficacy
The clinical trials as well as registries consistently confirm lower (target lesion) revascularization rates at follow-up with DES compared with BMS.9,16,18,20,21,33 However, the reduction of subsequent revascularization in registries was less than in clinical trials. The absolute reduction in target vessel revascularization at 4 years in a network meta-analysis of 38 clinical trials was about 12 vs. 2-4% in the Swedish SCAAR registry at 3 years.9,18 While the target lesion revascularization (TLR) rate in SCAAR (Swedish Coronary and Angioplasty Registry) did not reach 6% at 3 years in the DES arm,9 the network meta-analysis18 showed TLR rates up to 9% at 3 years with DES, a discrepancy that was even more apparent in patients treated with BMS and is most probably explained by systematic angiographic follow-up in many trials, and the lack of such angiography in the ‘real world’ registries. Data quality in the registries may be limited by the absence of event adjudication by blinded outcome assessors and lack of data query and verification, while patient and device selections are often operator dependent. In registries, patients treated with at least one DES are allocated into the DES cohort regardless of the number of simultaneously or previously implanted BMS, although clinical events in the mixed cohort may be related to either stent type. In many registries, the BMS group is a mixture of many different types of bare metal devices, with different outcomes. Similarly different DES are mixed while outcome may vary. This was illustrated from the SCAAR registry reporting restenosis rates per individual BMS types ranging from 7 to 11% at 3 years (Figure 1). For different DES 3 year restenosis rates varied between 3.3 and 4.6%. Finally, the interpretation of long-term follow-up data is hampered by cross-overs: patients who receive first one type of stent and then another type of stent at a later point of time, for example, for in-stent restenosis. Following the ‘intention to treat’ principle, all events were attributed to the first stent that was employed, while some new events may patho-physiologically be related to a second or third type of stent in that patient. In the original trial protocols, secondary stent thrombosis –stent thrombosis in a patient who had previously undergone target-lesion revascularization– was not considered to be a stent thrombosis. Consensus should be reached on how these events should be classified and reported.
Drug eluting stents and bare-metal stents in specific patient groups
In interventional cardiology as well as in other fields of medicine, post hoc subgroup analyses should be interpreted with caution. These may provide important directions for additional research, but the conclusions may only be accepted if these are very 375 strong, consistent among all studies, and based on plausible patho-physiology and experimental data. In general, the treatment effects in subgroups with limited numbers of patients and events are best estimated by the overall effects in the trials. Yet, a few subgroups should be discussed in this report.
ST-segment elevation myocardial infarction
Two-year follow-up of the STEMI cohort of the GRACE registry showed a lower in-hospital mortality in the DES cohort, a similar mortality rate from the time of discharge up to 6 months, but a significantly higher mortality rate from 6 months to 2 years in patients treated with DES (n=569) as compared with BMS (n=1729) (HR 6.69, 95% CI 2.05-21.8).34 Survival rates following DES or BMS were similar in both the overall population and in the non-STEMI group. A recent meta-analysis of trials including patients presenting with ST-segment elevation MI (eight trials-2786 patients) showed no difference in the hard clinical endpoints of death and MI in these high-risk patients. However, most of the trials had a follow-up limited to 1 year.35
Multivessel coronary artery disease
The recently presented 3 year results of the Arterial Revascularization Therapies Study (ARTS-II) reported the safety and efficacy
of percutaneous coronary intervention (PCI) using SES (n=607) as compared with the randomized surgical (n=605) and percutaneous arms (n=600) of ARTS-I for patients with multivessel coronary artery disease.36 The authors concluded that despite the higher clinical and angio-graphic risk profile of the ARTS-II population, the incidence of death/stroke/MI was significantly lower than in the ARTS-I PCI arm and similar to the ARTS-I coronary surgery (CABG) arm. Despite the significantly lower repeat intervention rates in ARTS-II as compared with ARTS-I PCI, CABG remained associated with the lowest re-intervention rates compared with both PCI groups. These findings were in line with the 3 year results of the Argentine Randomized Study Coronary Angioplasty vs. Coronary Bypass Surgery in Multiple Vessel Disease (ERACI-III trial; n=675) showing similar rates of death, stroke, and MI up to 3 years in patients treated with DES as compared with 415 either CABG or PCI with BMS and significantly lower repeat revascularization in the CABG group as compared with both PCI arms.37
Small vessels, long lesions, diabetics, and bypass grafts
Heterogeneity of the treatment effect was suggested by an analysis of the randomized BASKET-LATE trial,38 observational data from Ontario, Canada (n=16,498),21,39 and data from a continuous registry of all PCIs in Belgium (n=15,237). In these studies, the DES benefit was apparent particularly in small vessels, long lesions, diabetics, and bypass grafts. Among patients with diabetes, DES proved effective in reducing the need for revascularization in almost all lesion types and regardless of recent MI status. Among non-diabetic patients, the benefit of DES was more limited but was apparent in long lesions, small vessels and particularly when both adverse features co-existed. Also in the network meta-analysis, the number needed to treat to prevent TLR was lower in diabetic patients as compared with non-diabetic patients.18 Of note, in a meta-analysis of pivotal randomized controlled Cypher trials, significant heterogeneity in the treatment effects was found for patients with or without diabetes. The 4 year cumulative survival rates among patients without diabetes did not differ significantly between the two groups, while the survival rate for patients with diabetes was significantly lower in the SES group (P=0.008). Deaths from both cardiovascular and non-cardiovascular causes in diabetics (n=428) were more frequent in the SES group. In the subgroup of patients with diabetes, stent thrombosis more than 1 year after the procedure was adjudicated more frequently among the patients with SES than among those with BMS. Owing to the low number of events, these findings should be interpreted with caution; it does not appear that they adequately explain the observed difference in survival among patients with diabetes in the two groups.15 Conversely, in the larger scale network meta-analysis, no significant difference in all-cause mortality was observed in 3,679 patients with diabetes.18 Specific trials comparing different stents in these high-risk patient groups are warranted.
Conclusions, perspectives, and recommendations regarding efficacy and safety of DES
– Drug eluting stents, when compared with BMS as an initial treatment strategy, are associated with lower subsequent revascularization rates, but an excess risk of late stent thrombosis, which thus far, does not seem to impact on the occurrence of hard clinical endpoints like death and myocardial infarction up to 4 years.
– Thus far, the overall relative safety and efficacy of DES compared with BMS appears to be consistent across different groups of patients, albeit at various levels of absolute benefit and risk. The long-term safety and efficacy of DES in high-risk patient subsets such as diabetics and patients presenting with MI remains to be established.
– There are important differences between the various types of stents, with dissimilar mechanical and pharmacological properties and subsequent differences in clinical outcome: ‘Not all DES are equal, nor are BMS’.
– Interpretation of long-term follow-up data is hampered by cross-overs and mixed DES and BMS use: patients who receive first one type of stent and another type of stent at a later point in time, for example, for in-stent restenosis.
– These new findings add valuable information to the initial reports that have led to the creation of this Task Force and call for greater attention to scientific scrutiny and caution in the communication to the public of sensitive scientific information. However, the current conclusions regarding the safety and efficacy of DES should not divert the attention from the importance to continue development of less- or non-thrombogenic stents.
Health economical analyses
With respect to the discussions about effectiveness and risks of DES, and the higher costs of DES compared with BMS, health economical analyses have been initiated in different countries. Three analyses were presented, showing consistent results.
In Ontario, Canada, since 2003, the Ministry of Health & Long-Term Care has allocated a funding of $12 million for DES annually between the 12 Cardiac Care Centres in that province.39 Cost-effectiveness was analysed using data from 16,498 patients with at least 12 months follow-up receiving only BMS or DES from 1 December 2003-31 March 2005. Overall, the incremental cost-effectiveness ratios (ICERs) of DES vs. BMS were ‘fairly high’ (ICER ranging from $2630-for very long and narrow lesions- to $133,937-for short lesions in larger vessels). A significant benefit (reduction of repeat revascularization) at acceptable costs appeared in a limited group of patients with adverse lesion characteristics (long lesions, small vessels) and/or diabetes.
In its first appraisal of DES reported in October 2003, the English National Institute for Clinical Health and Excellence (N.I.C.E.) restricted the use of DES to small vessels (<3 mm) and long lesions (>15 mm) based on cost-effectiveness assessment using a QALY-based Incremental Cost-Effectiveness methodology. The expected use of DES at that time was ~30%, while in practice the usage increased up to >60% of patients in the UK. In July 2007, N.I.C.E. issued its second appraisal of DES in draft form and concluded that DES are not cost-effective in any population and cannot be recommended for patients with coronary artery disease. This recommendation was driven by a lower than expected need for repeat revascularization with BMS in unselected patient populations, markedly lower than in the randomized controlled trials, contrasting with the very high price of DES that did not fall as expected. Furthermore, the conclusion was driven by an increase in the cost differential between DES and BMS. This recommendation has been challenged by professional societies and industry alike, and has been the subject of extensive debate. It should be appreciated that the more successful the new technology, the more rapidly and further the price of the old technology that it is replacing is likely to fall. This may reduce the relative cost-effectiveness of the new technology, unless its price is similarly reduced. The appraisal committee reviewed their draft guidelines in October 2007 and N.I.C.E. subsequently made their 3 year review with the latest draft document issued in January 2008 (see Conclusions below) and did issue final guidelines in June 2008.
In the 18 month data presented from the BASKET trial, follow-up costs were similar for both DES and BMS and relatively low overall.38 Owing to higher stent costs, the use of DES was associated with an ICER of €64,732. In terms of clinical endpoints, DES proved most effective in high-risk patients and lesions, while the study showed no improved outcome in low-risk patients and lesions in terms of efficacy. Subgroup analyses revealed that at 18 months, the ICER for DES was favourable if the use was limited to high-risk patients with small vessel/bypass graft stenting (only one-third of the patients fall into these groups).
Ong et al40 evaluated the cost-effectiveness of SES in the Rapamycin Eluting Stent Evaluated at Rotterdam Cardiology Hospital (RESEARCH) registry and concluded that the SES was not cost-effective at 1 or 2 years follow-up when compared with BMS. The ICER per target vessel revascularization avoided would be 29,373 € at 1 year, and 22,267 € at 2 years in the total cohort. On the basis of these results, the calculated maximum cost-effective price after 1 year of follow-up is 1,336 € per SES for allcomers, or 1,023 € to achieve cost-neutrality in The Netherlands.
The findings of these health-economic analyses are consistent, indicating that DES are not cost-effective at the current price levels for most patients undergoing PCI for stable angina, while the use of DES can be cost-effective in a subset of patients at high risk for restenosis. It was discussed that DES would become cost-effective for most patients if the price-premium, relative to BMS, would not exceed 450 € (£300 Pounds Sterling). No cost-effectiveness data on the use of DES for the life-saving indications of PCI (unstable angina, NSTEMI, or STEMI) as compared with either BMS use or conventional treatment are currently available.
Conclusions, perspectives and recommendations on cost-effectiveness
– Cost-effectiveness analyses are essential to fully understand the value of BMS vs. DES and enlighten healthcare policies. However, careful interpretation is needed when analysing specific patient subsets derived from clinical trials that might not reflect the real world clinical practice.
– At the current price level, DES can be cost-effective when applied in high-risk patients. Alternatively, DES would be cost-effective in the majority of patients undergoing PCI at a price premium around 450 € (£300 Pounds Sterling) above the price of comparable BMS, or less. It is worth noting that the incremental cost-effectiveness ratio will be heavily influenced by the costs of both the new and old technologies. The more successful the new technology, the more rapidly and further the price of the old technology is likely to fall. This may reduce the relative cost-effectiveness of the new technology, unless its price is similarly reduced.
– Importantly, the available cost-effectiveness analyses do not pertain to high-risk patients such as NSTEMI and STEMI. Under those circumstances, PCI with BMS was shown to portend a survival benefit over non-interventional conventional therapy.1 It is unknown whether this survival benefit will be amplified by the use of DES.
UPDATE: NICE appraisal July 2008
Not discussed at the meeting
The current recommendation continues to support the use of DES in small vessels, <3 mm, and lesions, >15 mm, provided that the additional costs of the DES are £300 or less (see http://www.nice. org.uk/Guidance/TAI52).
The DES registration process
Drug-eluting stents are combination products that consist of a medical device with a medicinal substance as an integral part. DES were classified as class III medical devices and require a consultation with the Competent Authorities of the member states prior to CE certification by the Notified Body. Therefore, the Competent Authority will assess the clinical data related to quality, safety, and usefulness of the medicinal substance. The positive assessment report of the Competent Authority is the prerequisite for the certification of DES by the Notified Body.
In 2007, at least 19 different DES have received Conformité Européenne (CE) mark approval, while at the time of the conference only the first two DES (Cypher and Taxus) were approved by the US FDA for commercial use. Of note, the US FDA has recently given a positive review for the Endeavor and Xience-V DES. The German Society of Cardiology reviewed the data supporting 19 DES with CE mark, as reported in 76 randomized controlled trials. A distinction was made between studies with angiographic or primary clinical endpoints.41 The authors used a pre-defined decision tree to assess the level of evidence gained with the individual trials. Although the used score was not an internationally validated assessment tool for clinical studies and disregarded the fact that several studies were also powered for secondary endpoints, the authors concluded that only three, or at best five, out of the 19 CE-marked DES had adequate clinical documentation supporting their use. They concluded that there is significant heterogeneity in the requirements that are set for obtaining a CE-mark certificate.41 With the intention to harmonize the views and interests of the notified bodies on the one hand and the medical profession on the other hand, speakers from KEMA, BSI, TUV SUD Product Service, CETF, EMEA, and the FDA presented their views on the current approval process and their thoughts for modification of the process for assessment of next generation devices. There was agreement that novel stent technologies will be developed, and should be made available for patient use in Europe, but also that more extensive pre-marketing (pre-clinical and clinical) as well as post-marketing studies might be required. A balance should be found between pre-marketing and post-marketing evaluation with differential follow-up timescales, also accounting for the expected relatively short lifetime of the drug-device combination products. Additional guidance documents are required to achieve uniformity and consistency on the type of ‘short-term’ (pre-approval) data needed to receive CE Mark decision and ‘long-term’ (post-approval) data that are needed. Such documents are under development both by the Notified Bodies and by the EMEA. It was suggested by the clinical professionals that initial (CE mark) approval might be obtained based on assessment of the effects of a stent (DES or BMS) on coronary lesions (angiography, intravascular ultrasound), stent coverage by intima (optical coherence tomography or endo-thelial function studies), as well as 1 year clinical follow-up. In addition, post-market clinical follow-up should be considered for devices where identification of possible emerging risks and the evaluation of long-term safety and performance are critical. Careful attention should also be paid to differences in the populations enrolled in pre-market studies as compared with the actual use after release. Finally, the interplay between a device and long-term medical treatment, in particular, the duration, and the level of anti-platelet therapy should also be taken into consideration.
According to the EMEA, there are at present substantial differences in the amount and quality of submitted data. The authority emphasized the need for properly performed randomized controlled trials. While there is clearly a role for both patient-based endpoints such as death, MI, and repeat revascularization as well asfor lesion/device-based end-points such as late lumen loss, binary restenosis, and TLR, there is no consensus on how best to use both of these categories for trials at different stages in the development and approval processes. Furthermore, there are many unresolved issues with regard to the selection of comparators when evaluating new DES platforms. First, there are studies that should use surgery or medical therapy most appropriately as the standard of care for the control group. Secondly, if DES are compared with BMS, data presented from the SCAAR registry made the point that outcomes vary greatly depending on the brand of BMS selected (Figure 1).
In future studies, it may be relevant to compare new, investigational DES platforms to already approved
DES platforms in so-called ‘active control’ study designs. Yet, there is no specific requirement for either BMS or DES as a control device. When acceptable treatment alternatives exist, safety concerns can tip the balance in the risk-benefit assessment.
A proper randomization process with adequate concealment of allocation and blind adjudication of clinical endpoints is needed to avoid bias in the interpretation of data, but this can only be achieved in larger trials at relatively high costs. The balance between the need for robust pre-market evaluation and the speed of innovation in cardiovascular devices such as DES is a critical one that needs to be directly addressed to ensure patient safety and preserve the advance of medical technology.
Representatives of the US FDA confirmed that this organization also considers a two-staged approach: trials with a clinically relevant composite endpoint at 1 year (cardiac death, MI, and TLR) as well as assessment of death and MI and stent thrombosis over longer follow-up (at yearly intervals up to 5 years). It was suggested to use the recently established ARC-criteria for stent thrombosis (Figure 2). Furthermore, the actual use and possible interruption of adjunctive anti-platelet therapy should be registered. Both randomized clinical trials as well as properly organized registries of clinical practice might contribute to the post-marketing assessment of DES. In this process, it should be appreciated that new devices often are built on already existing platforms, polymers, or drugs. A completely new approval process may not be required for every small modification of an existing device, although this approach requires thorough characterization of DES through pre-clinical evaluation. Processes can be tailored for novel devices that incorporate already existing and approved platforms, polymers, or drugs. To reach a consensus on these issues, notified bodies indicated that they will encourage and dialogue with scientific societies, in an attempt to harmonize the relevant requirements coming from both a device-oriented and a pharmaceutical approach. There is a need for harmonization in interpretation and applying requirements for quality, safety and efficacy, and guidance to applicants and/or sponsors in planning the overall product development. Of note, the lifecycle of medicinal products and medical devices differ considerably; while new medicinal substances require time frames up to 15 years, the lifecycle of medical devices is shorter, ranging between 3 to 5 years.
A glimpse into future trial design
Currently, various innovative DES types are emerging and will become available in the coming years with the intention to avoid the current limitations of DES. A wide variety of modifications to the stent platform, coating, drugs, and eluting techniques are under investigation. Abolition of neointimal hyperplasia is no longer the ultimate goal, but rather the development of more biocompatible or bioabsorbable stents facilitating adequate endothelialization.42,43 At the same time, development of BMS continues, with new materials and geometry. Thus, future trials should not only compare new and established DES, but also new DES with new BMS.
Several proposals for large-scale randomized controlled trials are under development. These trials aim at an adequate power/sample size for hard clinical endpoints and are often conceived in a 2x2 factorial design to compare different stents and to simultaneously assess the optimal duration of (dual) anti-platelet therapy. For example, the PRACTICA trial might include ~12,000 all-comer patients, with a factorial design, randomizing first to treatment with either a DES or a BMS and subsequently for receiving 12 or 36 months of dual anti-platelet therapy. FIESTA has a similar factorial design, with a 5 year follow-up for death, MI, and repeat revascular-ization and 10 years follow-up for all-cause mortality. Only public funding will be sought for financing the trial. In the currently enrolling PROTECT trial, ~8,800 patients will be randomized to receive either a zotarolimus-eluting stent or an SES. Primary endpoint of this trial will be stent thrombosis at 3 years.
On a broader scale, the Cardiac Safety Critical Path Initiative, in which academics, industry, and regulatory authorities are joining, is initiating a programme focused on DES thrombosis and optimal dual anti-platelet therapy. The core of this programme is to develop a more formalized registration approach to concomitant evaluation of a drug and a device when there is an obligate interaction between the drug and the safety behaviour of the device.
Large-scale randomized trials (e.g. TRITON, OASIS 7, PLATO, and CHAMPION) are currently evaluatingthe pros and cons of newantiplatelet agents and will shed light on the balance between a possible reduction of adverse cardiac events by more intensive long-term anti-platelet therapy, perhaps at the costs of higher bleeding. Dual anti-platelet therapy with the P2Y12 receptor inhibitor clopidogrel and aspirin substantially reduces the risk of stent thrombosis. However, low response to clopidogrel is common and increases the risk for stent thrombosis. Clopidogrel is a pro-drug requiring conversion to an active metabolite resulting in a slow onset of effect. Multiple studies have demonstrated that the response to anti-platelet therapy with either clopidogrel and aspirin is highly variable when the responsiveness to clopidogrel is measured with ADP-induced aggregation. A low level of inhibition has been reported in 20-40% of the patients. Prasugrel-like clopidogrel is also a pro-drug, but with substantially more rapid onset
of P2Y12 inhibition and greater and more consistent platelet inhibition without the low responders seen with clopidogrel. Recently, the TRITON-TIMI 38 trial (n=13,608) compared prasugrel with clopidogrel when given before or during PCI and continued for 15 months in patients with acute coronary syndrome. Prasugrel reduced the risk of MI and halved the rate of stent thrombosis both with DES and with BMS. Prasugrel was, however, associated with an increased rate of major bleeding and transfusion, particularly in the elderly, in patients with
a low body weight, and in those with documented cerebrovascular disease.45 The net clinical benefit (death, MI, stroke, and major bleeding) favoured Prasugrel in most patients, except in the three subgroups mentioned above. Ongoing trials will reveal whether the new direct P2Y12 inhibitors, e.g. cangrelor, and AZD6140, or alternative doses of clopidogrel and prasugrel might overcome these limitations. Subsequently several of these agents should be tested at different treatment durations in combination with existing or new stents. Finally, the hypothesis has been developed that longer term dual anti-platelet therapy will not only reduce the risk for stent thrombosis but will also slow-down disease progression.
Conclusions, perspectives, and recommendations for trial design, registration processes, and anti-platelet therapy
– The EU Commission is asked to initiate the development of a unified guidance document for assessment of DES. The participation
of Competent Authorities of the Member States, Notified Bodies & EMEA and expert societies such as ESC and EAPCI is strongly encouraged in the development of such unified guidance for assessment. This standard should be homogeneous in Europe and flexible in order to allow new devices on the market. If appropriate, the guidance document might be written in consultation with the FDA.
– Randomized controlled trials pre-registration should strive to include all-comers which should be followed by large-scale all-comer registries to assess both the benefits and late complications. In view of the superior common goal of having complete unbiased longitudinal results after current and future DES, Industry, National Departments of Health and Scientific Societies should co-operate to implement prospective independent registries of all PCIs, ideally country-wide or continent-wide, with appropriate quality control and follow-up connected to official demographic registries. Attention should be given to the choice of comparator stents, since DES and BMS continue to develop.
– Both randomized trials and registries, reflecting the real world clinical practice, are valuable, however, they need to be put in their appropriate context. Rules for reporting events, endpoint definitions, and quality control need to be agreed upon and harmonized. Re-interventions should be categorized as clinically driven or non-clinically driven (e.g. angiographic follow-up, staged procedures, and evaluation prior to non-cardiac surgery).
– Market access should be based on efficacy as part of ‘usefulness’. Initial approval might be based on assessment of restenosis (angiography, IVUS) or neointima formation, and stent coverage (OCT, endothelial function) including 1-year clinical follow-up. These data should be followed by the assessment of death, MI, and stent thrombosis over longer follow-up (at yearly intervals up to 5 years).
– Randomized trials are required to assess the benefit, cost-effectiveness, and ‘optimal’ dose and duration of long-term (dual) anti-platelet therapy including the new more potent agents. Currently, the preferred design for prospective trials seems to be a 2x2 factorial design that incorporates a double randomization
to device and anti-platelet regimen.
– Data from clinical trials and registries should be in the public domain and academic investigators should have access to all relevant data for independent analyses and/or pooled analyses.
Appendix: Affiliations
1. Thoraxcenter, Erasmus MC, Rotterdam, The Netherlands
2. Cardiovascular Center, OLV Hospital, Aalst, Belgium
3. University of Liverpool, United Kingdom
4. KEMA Medical, Arnhem, München, Tel Aviv, Chalfont PA, Lafayette CA, Tokyo
5. Programs for Assessment of Technology in Health (PATH) Research Institute, St Joseph’s Healthcare Hamilton/Mc Master University, Hamilton, ON, Canada
6. TIMI Study Group, Cardiovascular Division, Brigham and Women’s Hospital, Harvard Medical School, Boston, MA, USA
7. University of Geneva, Geneva, Switzerland
8. Institut Cardiovasculaire Paris Sud, Massy, France
9. NHLI, Imperial College, London, United Kingdom
10. Clinique Pasteur, Toulouse, France
11. Heidelberg University, Ludwigshafen, Germany
12. Ospedali Riuniti di Bergamo, Bergamo, Italy
13. Trial Coordination Center, Department of Epidemiology, University Medical Center Groningen, The Netherlands
14. Department of Medical Sciences and Cardiology, Uppsala Clinical Research Center, University Hospital, Uppsala, Sweden
15. Division of Clinical Epidemiology and Biostatistics, Department of Social and Preventive Medicine, University of Bern, Bern, Switzerland
16. Deutsches Herzzentrum, Munich, Germany
17. TÜV SÜD Product Service GmbH, München, Germany
18. Department of Cardiology, Aarhus University Hospital, Skejby, Denmark
19. Duke University Medical Center/Duke Clinical Research Institute, Durham, NC, USA
20. CHU Sart-Tilman, Liège, Belgium
21. Departments of Cardiology, University Hospital, Basel, Switzerland
22. The London Chest Hospital, London, United Kingdom
23. Cardiology Practice and Hospital, Munich, Germany
24. Hôpital Bichat, Paris, France
25. BSI Product Services Healthcare
26. Bern University Hospital, Department of Cardiology, Bern, Switzerland
27. Biotronik GmbH & Co. KG, Berlin, Germany
28. Boston Scientific, Natick, Mass, USA
29. European Medicines Agency (EMEA), London, UK
30. Medpole, Neostent Corporation, Belgium
31. Center for Devices and Radiologic Health, United States Food and Drug Administration, Rockville, USA
32. Abbott Vascular, Santa Clara, CA, USA
33. Hexacath Corporation, France
34. Cordis Corporation, Johnson & Johnson Company, Miami Lakes, FL, USA
35. ClearStream Technologies Group plc, Wexford, Ireland
36. Sorin Biomedica, Saluggia, Italy
37. AFSAPS, Agence Française de Sécurité Sanitaire des Produits de Santé, Paris, France
38. Medtronic Vascular, Santa Rosa, CA, USA
39. Terumo Corporation, Tokyo, Japan
40. B Braun Medical BV, The Netherlands
