Introduction
A decade ago, the description of a primitive novel cell type capable of differentiating into cells expressing a mature endothelial cell phenotype and their capacity to incorporate into regions of active angiogenesis, witnessed the emergence of endothelial progenitor cell (EPC) biology1. The development and maturation of this new concept in vascular biology has resulted in numerous studies describing the role of EPCs in a myriad of disease states where abnormalities of the vasculature have been implicated. Thus, from pre-eclampsia to pulmonary hypertension, erythropoietin administration to erectile dysfunction and cancer to coronary disease the discovery of EPCs has added greatly to the understanding of basic pathophysiology. However, it is in the study of coronary artery disease where this paradigm shift has had greatest impact, not only regarding basic disease mechanisms, but in the rapid translation of these findings into a clinical context. The purpose of this review is to outline the current understanding of the EPC phenotypes and their relationship with risk factors for coronary disease. In addition, the potential problems of EPC dysfunction and its impact on percutaneous intervention will be appraised together with both pharmacological and stent based strategies to augment EPC number and function.
Defining the endothelial progenitor cell
Flow cytometry
EPCs have some basic characteristics of mature endothelial cells, including expression of markers such as CD34, a haematopoietic stem/progenitor cell marker and VEGFR2, one of the receptors for vascular endothelial growth factor (also known as Flk-1 and KDR). Early studies also described additional EPC characteristics such as incorporation of acetylated LDL and binding of lectins. Human haematopoietic stem cells and early EPCs also express another stem/progenitor cell marker which is not expressed by more differentiated cells and is therefore used to distinguish EPCs from more mature circulating endothelial cells that have sloughed off vessel walls. CD133, originally called AC133, is a 120-kDa transmembrane polypeptide, expressed on haematopoietic stem and progenitor cells from human bone marrow, foetal liver, and peripheral blood2. Thus, one generally accepted flow cytometric definition of EPC phenotype is the co-expression of CD34, CD133 and VEGFR2, but which do not express VE-cadherin and von Willebrand Factor. Cells with these characteristics are localised predominantly in the bone marrow. However, following various stimuli such as ischaemia or other forms of vascular injury, a more mature EPC phenotype appears in the peripheral circulation of adults, with lower levels of CD133 expression, but remaining strongly positive for CD34 and VEGFR2. It seems, therefore, that the loss of CD133 reflects the transformation of circulating EPCs into more mature endothelial-like cells. However, it is unclear at which time point the EPCs begin to lose CD133 expression – during their transmigration from the bone marrow into the systemic circulation or later, whilst in circulation. At present, it is unclear whether CD133 only represents a surface marker or has a functional activity involved in regulation of neovascularisation. The narrow phenotype of CD34/CD133/VEGRF2 positive cells may therefore represent only one subtype of putative EPCs and their comparative rarity (often <1 EPC per µl) in both the bone marrow and peripheral circulation make accurate flow cytometric measurement difficult.
Colony forming assays
In the context of these measurement difficulties, and also the requirement to assess not only EPC number but also function, the use of colony forming unit assays (CFU-EPC) has also been widely described1,3-5. These assays rely on the adherence properties of peripheral blood mononuclear cells to substrates such as fibronectin. Like many of the haematological colony forming assays, their interpretation relies upon operator identification and classification of the colony forming units. The direction of differentiation is governed by the choice of substrate and crucially the growth factors or serum supplements added to the baseline cell culture media. Achieving consistency in these characteristics has been the main determinant of success or not in EPC colony growth. However these problems have been largely overcome with the commercially sourced Endocult® EPC colony assay6 (Figure 1).

Figure 1. This is a picture of an EPC CFU-Hill colony (12.5 x magnification) grown in Endocult® medium, after five days of culture on a fibronectin-coated plate. The absolute size of a colony is approximately 0.35 mm, measured from the tip of a spindle shaped cell at the periphery across the body of the colony to the tip of a spindle shaped cell on the other side. Illustration provided by the courtesy of Stem Cell Technologies Inc.
The clinical relevance of these measurements however remains controversial and certain investigators have not been able to demonstrate a tight correlation between CD34 positive “EPCs” assessed by flow cytometry and colony forming ability7.
The role of CD14
Adding further to this phenotypic controversy is the experimental finding that putative CD34 negative angioblasts/progenitors have also been defined in vitro on the basis of their adhesion, growth, maturation and potential to augment (not necessarily by anatomical incorporation) angiogenesis in vivo8. Some groups would argue that peripheral blood derived EPCs which grow in culture are primarily derived from monocytic cells expressing CD14 rather than the flow cytometric markers described above9. This transdifferentiation potential of monocytic cells into cells with endothelial characteristics would suggest a possible common origin, namely a bone marrow-located haemangioblast10. Further evidence, however, would suggest that the original culture model of EPCs should be distinguished from the outgrowth endothelial cell population, which appears to grow exclusively from a CD14 negative fraction. Therefore, it is likely that in the broadest definition, the term EPC refers to a heterogeneous cell population comprising subpopulations that contribute to angiogenesis directly, by incorporation into new vessels, and indirectly, by positively regulating the angiogenic process11.
Novel approaches to characterise EPCs
For the reasons outlined above, it is apparent that new markers are urgently needed to reconcile the conflicting evidence in the literature, and to establish a more robust definition of EPCs as required for benchmarking and systematic analysis. Notably, the expression of individual marker proteins may be indicative but not sufficient to characterise stem cell-derived cells12. Instead, the ultimate phenotype of a cell is reflected in their instantaneous protein profile and proteomics, the global analysis of cellular proteins, offers an opportunity to progress towards a molecular classification of stem cell-derived cells13. While transcriptome analysis can be said to generate a cell specific signature14, it will not detail the true differences between stem cells and their progeny because of the importance of post-translational regulation and protein degradation15. The addition of a comparative analysis of protein expression patterns will add penetration and biologic information by distinguishing EPCs from other progenitors of the myeloid lineage and bridging the gap between their cellular plasticity and our current inability to characterise different EPC phenotypes.
Stent-induced vascular damage and re-endothelialisation
Stent implantation is an inherently thrombogenic procedure. Endothelial denudation manifests in a complex interaction between the stent struts and blood components, which includes the activation of platelets, the complement system and coagulation factors and ultimately the deposition of micro-thrombi on the stent surface. Restoration of a confluent and vasculo-protective endothelial monolayer is unpredictable and often patchy16. The regeneration of injured endothelium has several contributory mechanisms, including the proliferation and migration of neighbouring mature endothelial cells. The focal vascular injury incurred by percutaneous coronary intervention in stable patients is a sufficiently potent endogenous stimulus to mobilise bone-marrow derived EPC which also contribute to the repair mechanism17 (Figure 2).
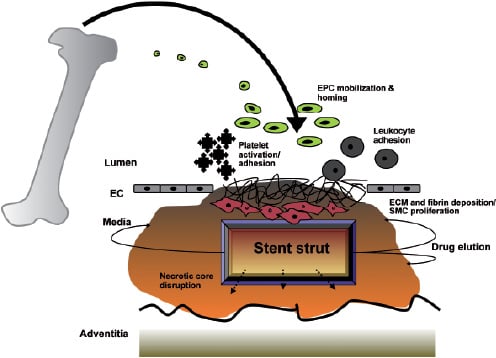
Figure 2. Schematic demonstrating that focal vascular injury incurred by percutaneous coronary intervention/stenting in patients is a potent endogenous stimulus facilitating a thrombogenic cascade and mobilisation of bone-marrow derived EPCs by complex and ill-defined signalling mechanisms. Circulating EPCs home to the region of injury and contribute to re-endothelialisation over the stented arterial segment.
Bone marrow transplantation experiments revealed that bone-marrow derived cells home to and contribute to the restoration of denuded arteries18. Another recent report demonstrated that infused peripheral blood monocyte-derived EPC home to bioprosthetic grafts and to balloon-injured carotid arteries, the latter being associated with a significant reduction in neointima deposition19. The signalling cascade mediating EPC mobilisation after an injurious stimulus is complex and not fully understood. Angiogenic cytokines including the prototypical vascular endothelial growth factor (VEGF), are thought to be involved20.
EPC function is dependent upon their concentration, mobilisation, homing capacity to regions of vascular damage and ability to incorporate into those sites of endothelial regeneration. It has been hypothesised that EPC serve a ‘surveillance’ role in the adult vasculature and mediate a low grade repair function for areas subjected to apoptosis secondary to ongoing exposure to cardiovascular risk factors21,22. Animal models have consistently shown the capacity of bone-marrow mobilised and circulating EPC to re-endothelialise regions of damaged vasculature and exert a vasculo-protective effect, but the true importance of this reparative mechanism in elderly patients predisposed to atherosclerosis is unknown. Patient-based studies have demonstrated the number of EPCs negatively correlate with coronary atherosclerotic risk burden, as defined by the combined Framingham risk score3,23, as well as cerebrovascular disease24. Recently, both diabetes and smoking have been shown to reduce numbers of EPCs and retard their proliferative and migratory response25,26. These defects in diabetics may in part explain their predisposition to endothelial dysfunction and subsequent vascular complications, particularly atherogenesis and impaired neovascularisation after ischaemic insults. Advancing age is a significant predictor of atherosclerotic disease and is associated with poorer outcomes, compared with younger individuals. Although the mechanism for age-related endothelial dysfunction is not known, impaired endothelial turnover and regenerative capacity is likely to be implicated27. Impaired survival, proliferative and migratory ability of EPC in elderly patients has also been linked with endothelial dysfunction28. Chronic renal impairment exerts a similarly deleterious influence on EPC functionality29,30. Interestingly, essential hypertension by itself appears to bear no correlation with EPC number and functional activity31. EPC activity has also recently been speculated to contribute as a surrogate risk factor to explain the gender differences in vascular disease predisposition, with both proliferative and migratory capacity being significantly greater in women32.
Late stent thrombosis and delayed re-endothelialisation
It is thus seen that impaired EPC activity may result in delayed re-endothelialisation and ‘wound’ healing post-stenting. The long-term success of coronary stent implantation, even in this modern era of DES, for flow-limiting atheromatous disease is limited by in-stent restenosis. The spectre of late and very late stent thrombosis has also recently emerged as a potential concern and is discussed below. The sheer volume of coronary interventions and their expanding indications ensure that late complications will continue to pose a significant burden33. ‘Conventional’ risk factors that predispose to restenosis have been extensively addressed elsewhere34 and takes into account patient co-morbidity (renal impairment, diabetes), anatomical considerations (longer lesion, complexity, target vessel diameter) and procedural inadequacy (stent strut malapposition). More recently, reduced numbers of circulating EPC and impaired adhesion capacity (fibronectin-binding) have been demonstrated in patients with in-stent restenosis35.
Drug elution and stent re-endothelialisation
The first generation of commonly used drug eluting stents (DES) incorporates sirolimus or paclitaxel on a polymer coated stainless steel platform. Sirolimus (rapamycin) is a macrolide antibiotic that binds to its cytosolic receptor FKBP12 and through inhibiting the down-regulation of the cyclin-dependent kinase inhibitor p27kip1, prevents G1 to S phase cell cycle progression thereby inhibiting smooth muscle cell migration and proliferation36. Paclitaxel is a derivatised diterpenoid that exerts an antineoplastic effect by interfering with cell microtubule function. It alters the dynamic equilibrium between microtubules and ß-tubulin by favouring the formation of abnormally stable microtubules37. This leads to the inhibition of smooth muscle cell division and migration, intracellular signalling, and protein secretion, which all rely on the rapid and efficient depolymerisation of microtubules. Significant (70%) reduction in neointimal proliferation is achieved at blood concentrations 100 times lower than antineoplastic levels38.
Delayed re-endothelialisation
Substantial evidence has emerged to support the hypothesis of delayed stent strut re-endothelialisation post implantation of DES. This possibility may have tremendous relevance in light of the currently debated controversy of the existence and clinical importance of late and very late stent thrombosis post DES implantation39. Direct visualisation by angioscopy of sirolimus eluting stents (SES), from several independent groups, provided evidence of incomplete neointimal coverage by EC 3-6 months post implantation, and this was associated with subclinical thrombus formation40,41. Furthermore, oral administration of everolimus in an animal model of iliac artery stenting, scanning microscopy demonstrated delayed endothelialisation of the stent surface42. Further supportive evidence from the same group is seen in an autopsy analysis of an individual who died of late stent thrombosis 18 months post SES deployment for unstable angina43. This showed aneurysmal dilatation of the stented segment, with histological evidence of a severe localised hypersensitivity reaction, consisting predominantly of T lymphocytes and eosinophils, associated with patchy areas of stent devoid of EC. A subsequent registry of 40 autopsy cases of presumed late stent thrombosis comparing the histopathological impact of both sirolimus and paclitaxel eluting stents with bare metal stents revealed poorer endothelialisation and persistent fibrin deposition44. Scanning electron microscopy data illustrates further compelling evidence of compromised re-endothelialisation of sirolimus and paclitaxel coated stents at both three days and 14 days post deployment in a porcine model (Figure 3).
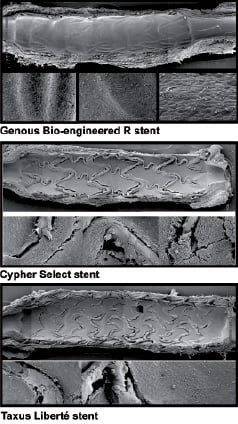
Figure 3. Scanning electron micrographs demonstrating the degree of luminal re-endothelialisation at 14 days after implantation in porcine model for Genous® Bio-engineered EPC capture stent (Orbus Neich; upper panel), sirolimus-eluting Cypher® stent (Cordis; middle panel) and paclitaxel-eluting Taxus Liberté® stent (Boston Scientific; lower panel). It can be seen that re-endothelialisation is almost complete with the EPC-capture stent, but there are patches of exposed stent strut visible with the sirolimus and paclitaxel-eluting stents. Illustration provided by the courtesy of Dr. Renu Virmani and Dr. Martin Leon.
Although there may be a temporal relationship of late stent thrombosis to cessation of antiplatelet therapy45 and the number of such patients appear small from the available registry data, the ability to identify specific factors which predispose to late stent thrombosis, or to identify those individuals who may require extended anti-platelet therapy would be of great value. It has become evident that the natural process of endothelial monolayer restoration (‘vascular healing’) after instrumentation and implantation of vascular devices such as stents, grafts or valves may take several months46. Accelerating stent endothelialisation to prevent both restenosis and late thrombosis and preservation of vascular homoeostasis, by recruiting circulating and/or tissue resident EPCs, is a novel and rational approach and has become a major challenge for vascular biologists and interventionists. Compounding this challenge is the fact that ageing and the associated accelerated senescence/apoptosis of the endothelial progenitor reservoir, impaired mobilisation and homing signals, may limit the regenerative capacity of EPCs and thus, their therapeutic potential, in the patient population who need it most.
Clinical applications and ongoing trials
Shifting of the vascular repair paradigm away from localised cytotoxic/cytostatic drug elution platforms to accelerated stent endothelialisation is an attractive concept. Regeneration of damaged endothelium is the fundamental process in vascular repair. Attempts to augment this process should reduce cardiovascular events associated with thrombosis, restenosis and hypertension47. Initial attempts to seed mature EC, using various delivery devices were frustrated by the rapid loss of seeded cells and in particular, the detachment of adhered cells to the vessel wall once antegrade coronary flow was resumed48. Attention has turned to the therapeutic value of EPCs and transplantation of EPCs for seeding of prosthetic grafts, stents or engineered blood vessels to create a bio-active EC layer has been successfully demonstrated19,49,50. The most recently attempted strategy utilised a magnetised stent, that after implantation into porcine coronary and femoral arteries, facilitated attachment and retention of superparamagnetically labelled EC51. After 24 hours, the delivered cells were retained in the presence of blood flow and also spread to the adjacent injured vessel wall.
Exogenous progenitor cell mobilisation from the bone marrow is a strategy used routinely by haematologists to facilitate recovery of the bone marrow after cancer chemotherapy. However, this approach has been hampered by the low concentration of EPC in the circulation and may also result in the non-specific capture of inflammatory cells. In this respect, non-invasive pre-treatment with pharmacological interventions such as G-CSF, statins, erythropoietin, oestrogen, and PPAR_ agonists all enhance the number of circulating EPCs and the potential to re-endothelialise damaged vasculature after revascularisation procedures52-56.
The HEALING first-in-man (FIM) study was a single centre safety evaluation of an EPC ‘capture’ strategy using stainless steel stents57. Sixteen patients with de novo coronary artery disease were treated with stents coated with anti-humanCD34 antibody. The nine-month composite major adverse cardiac and cerebrovascular events rate (MACCE) was 6.3% due to a symptom driven target lesion revascularisation (TLR) in a lone patient, and despite only one month of clopidogrel. The mean angiographic late luminal loss was 0.63±0.52 mm and the degree of stent volume obstruction was 27.2±20.9%.
The follow-up multicentre HEALING II study enrolled 63 patients, with procedural success achieved in all but one individual58. There was no subacute thrombosis, despite one month of dual antiplatelet therapy. The clinically driven TLR at six months was 6.3% with an overall MACE rate of 7.9%. Analysis of the circulating EPC levels stratified the patients into two subsets, those with a low and a normal level. It was noted that all restenotic and cardiac events were restricted to those patients with low EPC levels. Late luminal loss for the normal EPC group was 0.53±0.30 mm, but a binary restenosis rate of 0%. Conversely, those in the low EPC group incurred an in-stent late loss of 1.02±0.33 mm and a binary restenosis rate of 32%. Patients on statin therapy exhibited more than double the number of those that were not. This finding influenced the design of HEALING IIB, a multicentre, prospective, non-randomised study, which is currently recruiting approximately 90 patients with up to two de novo native coronary artery lesions. Patients are mandated to have two weeks of optimal statin therapy (80 mg atorvastatin) prior to the index procedure. The primary endpoint will be angiographic in-stent late loss at six months.
Statins (HMG CoA reductase inhibition) are well established to reduce cardiovascular risk and ischaemic events in vulnerable individuals and the numerous mechanisms whereby they exert their clinical benefit reflect the diverse molecular signalling pathway activated within numerous target cell types59. Most recently, they have been reported to promote neovascularisation of ischaemic tissue in normocholesterolaemic animals via activation of the Akt/eNOS pathway within endothelial cells60. Statins augmented peripheral blood CD34+/VEGFR2+ cells and their migratory capacity within four weeks of commencing therapy in patients with stable coronary artery disease61. The effect was deemed independent of cytokine stimulation, including VEGF and G-CSF, which have been shown to have a positive effect on EPC kinetics.
The results of the HEALING stent trials are awaited with interest. However theoretical risks remain and the non specific nature of the CD34 antibody raises the possibility that unwanted cell types, and not just EPCs, may also adhere to the stent struts. A recent report employing the use of anti-CD34 receptor coated arterio-venous grafts resulted in near complete endothelialisation. However, histological analysis of the grafts four weeks post implantation showed significantly higher restenosis rates in the CD34 coated grafts, compared to controls62. A recent study identified a new CD34-/ CD133+/VEGFR2+ subpopulation of EPC, which resulted in an even greater suppression of neointima formation and further augmented lumen circumference when transplanted to an animal model of carotid injury, compared with recipients of CD34+/CD133+/VEGFR2+ EPCs4. This data serves to reaffirm the widely acknowledged heterogeneity in EPC populations and interpretation of emerging clinical data needs to remain cautious.
Although the mechanisms underlying the homing and differentiation of EPCs at sites of injury is far from understood, integrins, a family of transmembrane adhesion receptors, are thought to play a crucial role63. CD34+ EPCs have been shown to express integrin binding motifs, including a cyclic Arg-Gly-Asp (cRGD). A novel polymer coated stainless steel stent, incorporating the cRGD peptide was designed as a novel ‘EPC capture’ stent64. There was a long term (three months) protective effect of the cRGD coated stents in a porcine coronary stenting model and merits further study.
Potential concerns with therapeutic use of EPCs
The clinical application of any new therapeutic modality is limited by several theoretical concerns, which need further study. One unresolved issue of harnessing an expanded circulating and/or resident EPC population for therapeutic restitution of the luminal endothelium is the consequence of unwanted vasculogenesis65. Neovascularisation in certain conditions, such as diabetes, is a ‘double-edged sword’. The formation of subintimal neovessels may potentially accelerate atherogenesis and facilitate plaque instability, and contribute to the restenotic process post stenting. Progenitor cells have been shown to contribute to the growth of arteriosclerotic lesions in a murine allograft model66. Increased rate of in-stent restenosis led to the withdrawal of the MAGIC randomised clinical trial using G-CSF for endogenous mobilisation of progenitor cells in patients who had a myocardial infarction67. The role of EPCs, however, in vessel wall pathology has not been extensively investigated and needs further clarification.
Secondly, EPC recruitment may lead to indiscriminate homing to sites of occult neoplasia, leading to vascularisation of dormant tumours68,69. The angiogenic effects of EPCs are mediated by growth factor secretion with therefore an unpredictable paracrine mechanism that would be also be difficult to quantify9. Although this concern has not been an issue in the angiogenesis clinical gene therapy trials for ischaemic heart disease70, the potential for this complication has not been rigorously investigated by EPC biologists.
Future directions
Apart from circulating EPCs, the value of non-bone marrow sources of progenitor cells needs further study. In particular, reports on local resident stem cells within the vasculature71 open exciting new avenues for novel therapeutic approaches if systemic or paracrine factors can be identified that regulate the activation and differentiation of these local stem cell populations. Further value in EPC therapy may also be gained by amalgamating transgenic technology, which provides the opportunity to use the cells as delivery vehicles of therapeutic genes. Vector mediated overexpression of genes with antithrombotic, vasodilatory or antiproliferative properties to enhance the function of implanted cells to prevent thrombosis and restenosis are in the early stages of evaluation. Overexpression of nitric oxide synthase (eNOS) gene within transplanted autologous EPCs in injured vessels enhanced the vasculo-protective properties of the reconstituted endothelium, leading to a greater inhibition of neointimal hyperplasia, compared to vessels treated with a control vector72. Endovascular implantation of cell-coated stents remains a possible goal; improved stent coverage of EC was recently achieved by seeding of ECs overexpressing VEGF73.
Conclusions
The paradigm of EPC biology and its fundamental relevance to cardiovascular health and disease is firmly established. Progress in the field is rapid and difficulties that plagued the early animal studies such as concordance amongst investigators on the precise phenotype of the putative EPC may be aided by novel approaches such as proteomic analysis. Pathophysiological sequelae in the atherosclerotic vessel wall, in terms of both the atherogenic process and response to vascular injury including stenting, result in the mobilisation of bone-marrow derived and vessel wall resident EPCs as part of an endogenous vascular repair mechanism. Evidence is emerging that the observed late and very late drug eluting stent complications (thrombosis, in-stent restenosis) may be attributable, in part, to inadequate stent re-endothelialisation. Accelerating stent re-endothelialisation is being evaluated in the HEALING stent trial program, using an EPC capture stent, which has shown early promise and continues to gain momentum with the actively recruiting HEALING IIB trial. Embracing the role that EPCs play in vascular homoeostasis and repair and an acknowledgement of their increasingly important therapeutic potential is a true step forward for bridging the divide between molecular therapeutics and interventional cardiology.
