Abstract
Aims: To introduce the interested reader to the concepts of the Clinical Event Committee (CEC) work process with a focus on the adjudication of major endpoints in contemporary coronary stent trials.
Methods and results: Endpoint adjudication by independent Clinical Events Committees (CEC) is critical to ensure the generation and recording of quality data in clinical outcome trials. CEC adjudication provides a standard, systematic and unbiased assessment of endpoints. For trials with relatively long-term clinical endpoints that span geographic regions and include diverse clinical presentations and practice patterns, this poses specific challenges. The recently published RESOLUTE All Comer coronary stent trial is used to illustrate some aspects of the CEC process.
Conclusions: Understanding the CEC review process is important to guide the design of future trials and allow meaningful comparisons of event rates among trials.
Introduction
In order to evaluate the effects of a particular treatment strategy on mortality and major morbidity within a disease entity, large, truly global clinical trial programmes with relatively long-term clinical endpoints have become the accepted standard. The critical challenge in the conduct of endpoint trials relates to the definition, collection and accurate assessment of endpoint data in a consistent timely manner.
In the context of coronary stent research, the need for uniform endpoint definitions and the harmonisation of clinical event adjudication has been addressed by the Academic Research Consortium (ARC) group.1,2 These definitions were intended to be applied to relatively homogeneous populations with stable presentations of coronary disease. An important challenge in this process is to adapt the initial definitions, while maintaining accuracy and consistency across geographic areas and over the long course of the study. This may prove even more important while addressing minimally selected populations, including complex patients, reflecting routine clinical practice.3 To serve this purpose a growing number of coronary device trials rely on the adjudication of safety and efficacy endpoints by independent Clinical Event Committees (CECs), also known as Clinical Endpoint Committees. Event and data adjudication by the CEC is a critical factor in generating and recording quality results, facilitating acceptance of the trial results by regulatory bodies, as well as the academic and medical communities.
It is the responsibility of the CEC to review all relevant and available data and provide an independent, blinded determination of trial specific endpoints or events utilising pre-specified criteria and considering the clinical scenario. CECs must be rigorous in their analysis of the data to ensure the highest potential value of the science. Since there are no accepted standards of what constitutes conclusive evidence, the process by which committee members arrive at their decisions must be made fully transparent. The quality of their work may be assessed on an on-going basis throughout the clinical trial and/or trial programme via internal and/or external validation.
In this article we will highlight the key concepts of the CEC process with a focus on the adjudication of major endpoints in coronary stent trials. The recently published RESOLUTE All Comer coronary stent trial (Resolute-AC) is used to illustrate some aspects of the CEC process.4,5
CEC structure and process
A CEC should comprise three or more practising physicians with particular expertise in the subject of the research. Trial specific characteristics, such as the trial size, estimated event rates and number of endpoints, will dictate the number of committee members required. They should not be otherwise involved in the study (e.g., as investigators, members of the Data Safety Monitoring Board [DSMB], or Steering Committee) and should not have a financial interest in the sponsor. Ideally, the choice of CEC members and the operational aspects of the CEC process should be delegated by the sponsor to an independent academic research organisation (ARO). Where these tasks are undertaken by the sponsor, it is essential that pre-specified procedures, documented in the CEC charter, exist to ensure that the independence of the CEC is not compromised.
The different key components of the CEC process are depicted in Figure 1. They should be pre-specified in the CEC charter as a part of the study protocol. The CEC charter should provide a detailed description of the structure, the membership, and the role and responsibilities of the CEC. The workflow and working procedures, including all administrative as well as methodological aspects of the CEC work, should also be clearly outlined. The endpoints should be carefully defined and the the strictest possible criteria must be set. The criteria should also be simple in order to avoid misinterpretations.
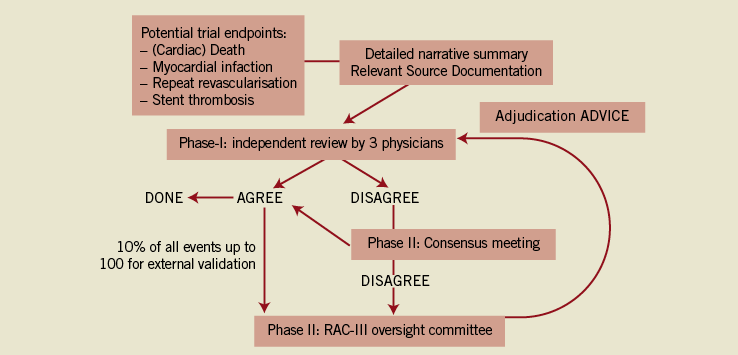
Figure 1. The clinical event committee adjudication process.
The CEC must commit to conducting a paced and timely review of cases identified as having potential endpoints. This process should also allow for the generation of interim data that may be reviewed by DSMBs and the Steering Committee.6 The total number of adjudicating members present at any meeting should be two or more and preferably odd in number to come to the final adjudication. In the Resolute-AC trial, both a web-based, independent review and the (face-to-face) consensus meeting methods were used. Given the complex patient population and the fact that several “teams” of reviewers were working in parallel, all potential events were first reviewed by three independent reviewers using a web-based system. When there was consensus agreement, then the adjudication was considered final. When there was disagreement, the event was discussed at a face-to-face consensus meeting, during which the CEC panel was asked to deliberate until every effort had been made to reach a unanimous decision whenever possible. In case of disagreement, the decision was by majority vote (>50%) of voting members present. For each case, a brief written justification of the reasons for a decision was provided. This allowed internal review of cases that had been decided by majority vote, to ensure consistency among CEC meetings with different members present.
Suspected endpoint events (“event triggers”) are identified by using reports developed within the various study databases. Suspected endpoint events can be reported by the investigators at the clinical sites, identified through programmed queries based on triggers from the case report forms and other study data (e.g., angiographic or electrocardiographic core laboratory reports, specific patterns of release of biomarkers of myocardial damage) or following careful review of source data by the CEC. Figure 2 shows the data source for the event triggers in Resolute-AC and their relationship to the final reported outcome. The CEC reviews endpoint triggers and all available data from case report forms and any accompanying source documentation as needed. This process is strongly dependent on the information provided by an investigator, including endpoint event reporting. This information should be complete, accurate and valid. Ideally, every item of data that appears in a case report form (CRF) should be documented somewhere else to allow verification, audit and reconstruction (see also: “data reporting and source data verification”). Source documentation packets should be blinded to both subject identification and treatment assignment. Clinical interpretation (from the Clinical Reviewer) and judgement (from the CEC) are important to cluster multiple event triggers correctly into one event (e.g., acute myocardial infarction [AMI] from multiple cardiac marker test results and ECGs, stent thrombosis adjudicated from AMI and from cardiac death). One final potential role of the CEC is to identify “problem” sites where there is a pattern of deviation from study mandated protocols (e.g., failure to collect the required biomarker data or systematic performance of follow-up angiography where there is no clear clinical indication).
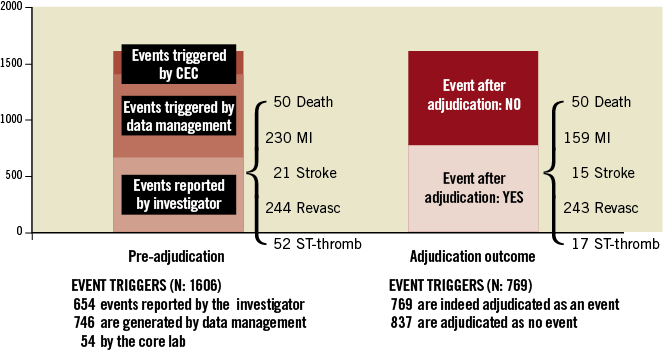
Figure 2. Clinical event triggers in the Resolute-AC trial (RAC). The left panel illustrates the source for the suspected event (“triggers”), the right panel the final event adjudication.
Within a study or clinical trial programme (set of trials evaluating the same product) quality control (QC) procedures may be implemented to identify potential problems and areas for improvement in the CEC adjudication process. Given the consensus review method and high level of unanimous decisions, a formal intra- and inter-observer variability assessment at the individual CEC team level is not usually planned. Yet, a predefined percentage of randomly selected, previously adjudicated events may be resubmitted to other members within the same CEC committee (internal validation) or to another, independent, overseeing CEC committee (external validation). The latter will integrate a QC of CEC operations (e.g., the workflow and working procedures).
Event adjudication beyond any reasonable doubt
The clinical adjudication process by itself should be highly specific, accurate and consistent, mainly driven by the application of pre-specified criteria for event definitions applied within a specific scenario. However, in cases where the minimal required data for event adjudication is permanently missing, event adjudication is based on interpretation of the available data, using a worst case scenario tempered by clinical judgement and, by consequence, more prone to variability.
Endpoint definitions
The effort of ARC with the objectives of establishing consistency among endpoint definitions and providing consensus recommendations for stent investigations in stable patients with de novo coronary lesions was pursued and extended towards non-selected patient populations.4,5,7,8. A critical appraisal of the ARC endpoint definitions is beyond the scope of this paper and provided in a separate paper.9 We will focus on those elements important to event adjudication and trial design in contemporary coronary stent trials including complex patient populations.
The ARC consensus proposed two composite endpoints for coronary device trials, one that is oriented towards procedure and device related risks and benefits (cardiac death, myocardial infarction attributed to the target vessel, clinically driven target lesion revascularisation), and a broader one that is oriented towards the overall patient-oriented clinical outcome. The emphasis should be on the latter.
In coronary stent studies with relatively long-term clinical endpoints, progression of coronary artery disease involving remote areas may become relevant. For protocols involving multivessel, multi-segment stenting, it may be problematic to attribute the event (e.g., myocardial infarction, stent thrombosis) directly to the pre-specified target lesion and study stent.
When target-vessel and remote area events cannot be differentiated based on the available source documentation (e.g., ECG, coronary angiogram, autopsy findings) these events should be attributed to the study device.
Death
ARC considered all-cause mortality the most unbiased method for reporting deaths in a clinical trial, even though it may be less specific than deaths adjudicated as due to cardiac aetiologies. All-cause death can be readily ascertained with minimal bias or need for adjudication. For situations when attribution to cardiac versus non-cardiac causes is desired, such as medium to long-term follow-up studies, ARC proposed a conservative approach considering the worst case scenario. Sudden or unexpected death even in patients with coexisting potentially fatal non-cardiac disease status (e.g., cancer or sepsis) should be classified as cardiac, including the “unknown”, unless history related to the non-cardiac diagnosis indicates death was imminent. Clinical autopsies may help to establish the true cause of death and should be strongly recommended.10 In Resolute-AC an additional category for classification of death, namely “unexplained” was available to the CEC, which defaulted to cardiac death for statistical analyses. This allowed the CEC to indicate that, were further source documentation provided, the cause could be reclassified as “non-cardiac” or, in the case of cardiac death within one month of the index procedure, further source documentation could clearly rule out a probable stent thrombosis. Commonly encountered situations were, firstly, death in patients undergoing primary percutaneous coronary intervention (PCI) at index, who were unwell but apparently stable in the subsequent days, but then died in circumstances where the source documentation did not establish a clear cause and did not allow the CEC to determine whether death was “expected”. A second scenario, increasingly common after twelve months, was death where information from family members suggested the cause was non-cardiac (e.g., cancer) but where no source documentation was provided.
Myocardial infarction
The diagnosis of acute, evolving or recent MI requires, in the absence of pathologic confirmation, findings of a typical rise and/or fall of a biomarker of myocardial necrosis, in conjunction with clinical signs or symptoms that the cause of myocardial damage is ischaemia.11
After careful consideration, the ARC endorsed the joint ESC/ACCF/AHA/WHF (European Society of Cardiology, American College of Cardiology, American Heart Association and World Heart Foundation) Task Force 2007 universal definition of myocardial infarction for consistent application across investigational studies; however, they advised the use of creatine kinase-myocardial band (CK-MB) mass over cardiac specific troponin (cTn) as the primary marker of myocardial necrosis.2,12 It has been contended that cTn may be overly sensitive, increasing the overall rates of PMI and exaggerating differences in clinical outcomes between treatments which are neither clinically nor prognostically relevant.13-16 This may be even more of an issue in contemporary all-comer coronary stent trials allowing the unrestricted use of DES.4-5 Moreover, and as illustrated in Figure 3, definition instability may be encountered while considering patients with acute coronary syndromes with or without on-going myocardial ischaemia at the time of the index procedure and/or while being confronted with missing biomarker sets.
Reinfarction following primary PCI, in patients with a presenting myocardial infarction, can only be assessed if there is a clear indication that the cardiac biomarker sample values were falling following the index event and were then rising again in the immediate periprocedural period of 48 hours (Figure 3). Otherwise, there would be insufficient biomarker data to adjudicate a PMI. A 20% increase is considered significant, however, this value must exceed the appropriate threshold according to the timing of the event (post PCI or post coronary artery bypass grafting). Of note, the latter requirement was not included in the index publication.12 In the case of reinfarction following elective PCI in patients with a recent MI (up to 48 hours, thrombolysed or not) and only one biomarker set available pre-procedure, one may assume biomarkers to be falling, although this cannot be established with certainty. Here, judgement may be guided by the clinical scenario (e.g., timing of the procedure) and careful evaluation of serial 12-lead ECG and/or angiographic findings (e.g., occluded vessel, thrombus, or loss of side branch). The Resolute-AC Steering Committee decided to report endpoints both according to the 2007 universal definition of MI and also the modified historical WHO MI definition extended also to include acute coronary syndrome (ACS) patients.4,5,16 For MI due to PCI, it is important to distinguish events defined by a threshold level of enzyme or biomarker elevation where the degree of elevation has a proven relationship to other more meaningful clinical outcomes.17-19 The clinical relevance of the currently set thresholds levels of enzyme or biomarker elevation remains to be established.
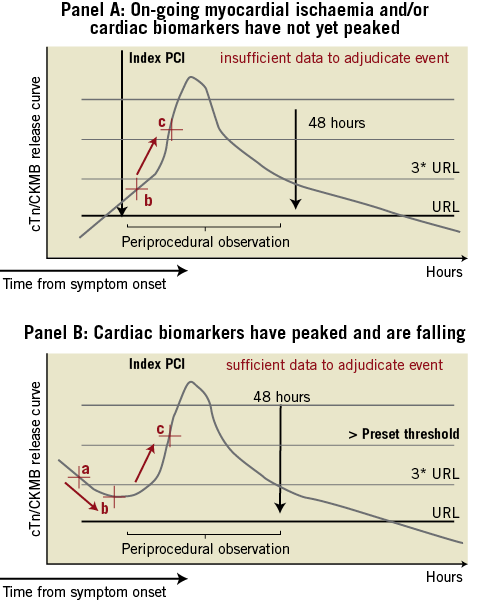
Figure 3. ARC MI definition instability. ARC MI definition instability may be encountered in acute coronary syndromes with (scenario A) or without (scenario B) on-going myocardial ischaemia. *Scenario A: Patients with on-going signs and/or symptoms consistent with myocardial ischaemia and the cardiac biomarkers are not yet elevated and/or have not reached peak values. Two cardiac biomarker values were sampled post PCI: the first six hours post PCI (b), the second three to six hours later. There are insufficient data to adjudicate a new MI event following primary PCI considering the cardiac biomarkers. *Scenario B: cardiac biomarkers have reached peak values and are falling (cardiac biomarker sample values a-b). Any significant rise starting following the primary PCI will constitute a reinfarction (MI extension) (cardiac biomarker sample value c). The same criteria should be applied beyond 48 hours if the biomarker level has not yet returned below the upper reference limit. The criteria for spontaneous MI does not apply. URL denotes upper reference limit for the cardiac biomarkers used to detect myocardial injury.
Myocardial infarction after the predefined immediate periprocedural period of 48 hours (72 hours for coronary artery bypass grafting), and after the cardiac biomarkers used to detect myocardial injury have returned below the 99th percentile of upper reference value in a normal reference population, may be secondary to late stent complications or progression of the native disease (“spontaneous”). ARC endorsed the 2007 universal definition of MI criterion for MI of any elevation of cTn above the upper range limit.
With sudden, unexpected cardiac death, involving cardiac arrest, especially when accompanied by clinical signs or symptoms of myocardial ischaemia, the CEC may adjudicate MI considering the clinical scenario including pathological findings, and new wall motion abnormalities on noninvasive imaging.
Reintervention
Approximately 50% of all repeat revascularisations in the RAC study were clinically justified target lesion revascularisation (TLR).4,5 The ARC acknowledged the importance of clear and consistent definition of TLR to an understanding of variations in drug-eluting stent (DES) clinical effectiveness in reducing restenosis, whether across different patient populations, lesion categories or the tested devices themselves.2 The clinical justification for TLR includes two fundamental components: the luminal measurement (angiographic component) and the clinical context. The quantitative pre-adjudication of coronary angiograms for lesion localisation, severity and visual intracoronary thrombus by an independent quantitative coronary angiographic (QCA) core laboratory should contribute to the quality of the process. The CEC is asked to judge the clinical justification in intermediate severity stenoses (50-70%) by assessing the presence of symptoms consistent with myocardial ischaemia in conjunction with the site-reported results of noninvasive functional testing. Protocol deviations with systematic follow-up angiography seem to be the norm in some geographic areas. In such cases, a cardiac basis for site-reported “symptoms” (if any) and their potential relation to the target vessel (in the presence of other untreated disease) mean that even “intermediate” restenoses are classified by default as clinically driven. While such events will not influence the results of a randomised DES trial, they have negative implications for the interventional community and for comparisons with surgery. An invasive functional (physiological) assessment by coronary pressure-derived fractional flow reserve (FFR) may provide important added value for intermediate lesions. However, where this is not pre-specified by protocol, for all such lesions, and subject to independent core lab validation, its value is dubious.
Stent thrombosis
Stent thrombosis is a catastrophic complication of coronary stenting, presenting as sudden death or nonfatal myocardial infarction (MI) in almost all cases. ST should not be considered an endpoint outside the overall clinical context. Concerns remain surrounding those events occurring beyond one year that may present as unexplained death. Limiting the ARC possible ST criteria to include death beyond 30 days due only to sudden death acute ischaemia may provide the best estimate of late and very late stent thrombosis rates.10
Data collection and monitoring
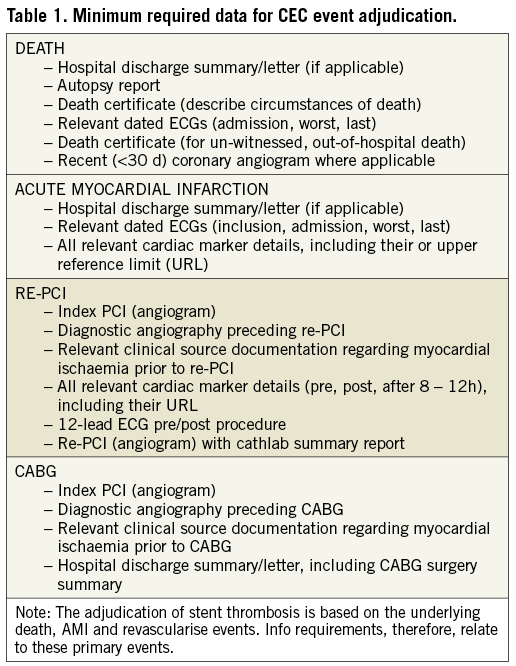
The strategy used to identify and adjudicate endpoint events is one of many factors to be considered when comparing event rates between clinical studies. Only a minority of coronary stent trials detail the techniques relating to the ascertainment of and data collection for hospitalised and non-hospitalised events. Table 1 lists the minimum required data (coronary angiograms, autopsy reports, etc.) for CEC event adjudication for major outcome events in the Resolute-AC trial. For periprocedural MI, an analysable set of cardiac biomarkers (either CK, CK-MB or cTn) consisted of a baseline value and at least two subsequent measurements within 48 hours post procedure at ≥4 hour intervals. In patients with an inclusion MI, a minimum of three different samples is required (Figure 3, scenario B). As indicated earlier, clinical autopsies are vital to establish the true cause of death and should be strongly encouraged. This issue becomes even more critical in the late follow-up period when there may be a higher frequency of death and details are more likely to be inadequate for specifying a cause.10
In quantitative clinical research, the quality of CEC adjudication is highly dependent on the completeness and detail in individual patient accounts, including protocol-driven test results (laboratory examinations, standard 12-lead ECG). The potential underreporting of adverse events is an important limitation of large multicentre registries where data monitoring is only provided for a fraction of all events. An audit of a clinical trial provides the research sponsor with independent appraisal of the quality and completeness of the data generated by the trial. Although auditing alone cannot transform a poorly planned, executed, monitored, or analysed trial into a credible one, an active clinical trial audit programme will point out potential problem areas early, so that solutions can be put in place before it is too late. In addition to providing objective information about processes, audits can prevent future errors by identifying problematic work patterns or behaviours. Anticipation of an audit may be an important quality assurance mechanism, providing sites with an additional incentive to maintain data quality.
Conflict of interest statement
The authors (PV, EMF, RM, DC) are all members of the Academic Research Consortium and have contributed equally to this document, drawing on their academic and clinical experience. All are involved in research work in collaboration with industry (i.e., the Medtronic Resolute coronary stent study programme), governmental or private health providers.
