Abstract
Aims: There have been recent concerns regarding the long-term safety of the first generation of drug-eluting stents, which utilised a permanent polymer coating for drug delivery. SERIES I is a prospective, non-randomised, first-in-man open label study with the biodegradable polymer-based Supralimus® sirolimus eluting stent (Sahajanand Medical Technologies Pvt. Ltd, India) for the treatment of patients with coronary artery lesions.
Methods and results: One hundred patients were treated with 126 Supralimus® stents (mean lesion length 10.5±4.3 mm, mean reference vessel diameter 2.66±0.62 mm). The pre-specified primary endpoint was angiographic binary in-stent restenosis at six months. Secondary endpoints were device-orientated major adverse clinical events (MACE; defined as a composite of cardiac death, nonfatal myocardial infarction [Q-wave and Non-Q wave], or clinically-justified target vessel revascularisation) at 30 days, nine months and 30 months. Angiographic follow-up in a pre-specified subgroup of 60 patients at six months showed binary angiographic restenosis rates of 0% (in-stent) and 1.7% (in-segment). The in-stent late loss was 0.09±0.37 mm. MACE rates were 0% after one month, 6% at 9-month follow-up and 7% after 30 months follow-up.
Conclusions: The biodegradable-polymer-based sirolimus-eluting stent (Supralimus®) is effective in inhibiting neointimal hyperplasia.
Introduction
Although the first generation of drug eluting stents (DES) have drastically reduced rates of restenosis and revascularisation1-5, concerns persist regarding their long-term safety6-8. The presence of a permanent polymer coating may contribute to stent thrombosis as a result of delayed healing and a hypersensitivity reaction in some cases9-14. The sirolimus-eluting stent currently approved for clinical practice (Cypher, Cordis, Warren, NJ, USA) uses a non-erodable polymer (polyethylene-co-vinyl acetate [PEVA] and poly n-butyl methacrylate [PBMA]), which may be responsible for the eosinophilic infiltration of the arterial wall seen in animal studies; the hypersensitivity reaction occurs after the complete release of drug, suggesting that the polymer may be the cause15. To address this issue, a new generation of DES is currently under development, incorporating biodegradable, biocompatible polymers as vehicles for drug delivery. Full degradation of these polymers into carbon dioxide and water ensures complete drug release, leaving a residual bare metal stent16-19. We report on the angiographic and long-term clinical follow-up of a novel biodegradable-polymer-coated sirolimus-eluting stent.
Methods
The Supralimus® stent consists of an established balloon-expandable 316L stainless steel slotted-tube stent platform (Matrix, Sahajanand Medical Technologies Pvt. Ltd., India) with two layers of degradable polymer coating: the base layer consists of 1.4 mcg/mm2 sirolimus incorporated into a biodegradable polymer matrix consisting of PLLA (Poly L-Lactic acid), PLGA (50/50 Poly DL-Lactide-co-Glycolide) and PVP (Polyvinyl Pyrrolidone), with a drug:polymer ratio of 35:65. The outer protective layer containing only PVP prevents premature drug release and is completely removed within two hours after implantation.
Following removal of this protective layer, an early burst phase releases 50% of the drug within the first seven days to inhibit the inflammatory response and smooth muscle cell migration and proliferation. The remaining 50% of sirolimus is released within 41 days; thus in an average of 48 days, the total drug content is released from the stent surface, as demonstrated in Figure 1.
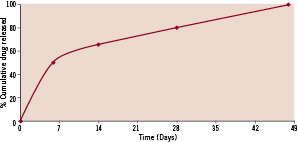
Figure 1. Cumulative in vitro release profiles of sirolimus from Supralimus® stents.
Porcine studies of the Supralimus® stent have demonstrated partial and complete endothelialisation after eight and 28 weeks respectively, with no evidence of hypersensitivity. The polymer and stent platform have both already been tested in humans, albeit incorporating a different anti-restenotic drug, with no undue concerns regarding safety17.
The interventional procedures were performed according to current standard clinical practice20. All patients were pretreated with aspirin and either clopidogrel or ticlopidine. Dual antiplatelet therapy was maintained indefinitely after the procedure. Heparin was administered to maintain an activated clotting time of greater than 250 secs.
One hundred patients were enrolled. Inclusion criteria were stable or unstable angina; a reference vessel diameter between 2.5 and 4.0 mm and lesion length: < 25 mm, which could be covered by a single Supralimus® stent (available stent sizes were from 2.5-4.0 mm diameter and 11-33 mm in length). Exclusion criteria included a platelet count < 100,000 cells/mm3 or > 700,000 cells/mm3; WBC of < 3,000 cells/mm3; documented or suspected liver disease (including laboratory evidence of hepatitis); recipients of heart transplants; known allergy to aspirin, clopidogrel, ticlopidine, heparin or stainless steel; ST elevation myocardial infarction within past 24 hours; presence of chronic renal failure (creatinine > 2.5 mg/dl); advanced malignancy; scheduled for major non-cardiac surgery within six months of PCI.
The pre-specified primary endpoint was binary angiographic in-stent restenosis. Secondary endpoints were composite major adverse clinical events (MACE), defined as a composite of cardiac death, non-fatal myocardial infarction (Q-wave and Non-Q wave), or clinically justified target vessel revascularisation at 30 days, nine months and 30 months.
Angiographic follow up was proposed to all patients, but was carried out only on those consecutive patients who consented, numbering 60 in total.
Quantitative coronary angiography (QCA) was performed by an independent core laboratory (Cardialysis BV, Rotterdam, The Netherlands) using the CAAS II analysis software (Pie Medical BV, Maastricht, The Netherlands). The stented segment refers to the stent and 5 mm proximal and distal to the stent edges. The following data were obtained: minimum lumen diameter (MLD), interpolated reference vessel diameter and percentage diameter stenosis (DS). Binary restenosis was defined as a DS ≥50% at follow-up angiography. Late loss was defined as the difference between MLD post-procedure and MLD at follow-up. Video-densitometric QCA was also performed to obtain the reference vessel area (RVA), minimum lumen area (MLA) and percentage area stenosis21-23.
Results
On hundred and twenty-six Supralimus® stents (diameter range 2.5-4.0 mm, length range 11-33 mm) were implanted in 100 patients (mean 1.26 stents per patient). The PCI procedure was successful in all cases. The baseline patient and lesion characteristics are shown in Tables 1 and 2.

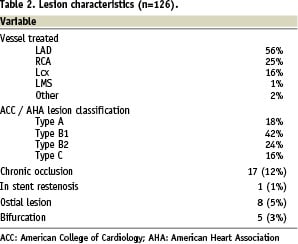
The mean lesion length was 10.5±4.30 mm. The mean stent length and diameter were 18.95±7.58 mm and 2.95±0.44 mm respectively. Angiographic follow-up after six months was available in 59 out of 60 scheduled patients. These results are shown in Table 3; the rates of angiographic binary in-stent and in-segment restenosis were 0% and 1.7% respectively.
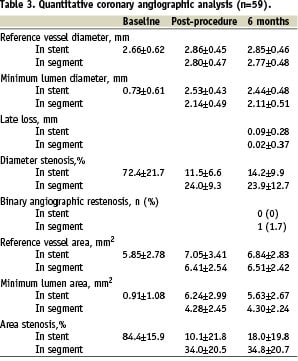
All the patients were clinically followed up at 30 days, six, nine, 24 and 30 months. Follow-up data was available in all the living patients, up to the pre-specified trial end point of 30 months. There were no in-hospital complications and no adverse clinical events at 30 days follow-up. After nine months, the composite MACE rate was 6%. The long-term clinical follow-up is shown in Table 3 and Figure 2.
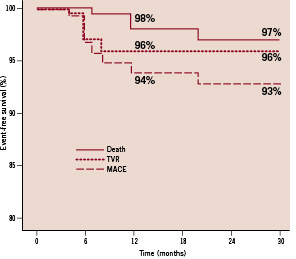
Figure 2. Kaplan-Meier estimates of clinical events.
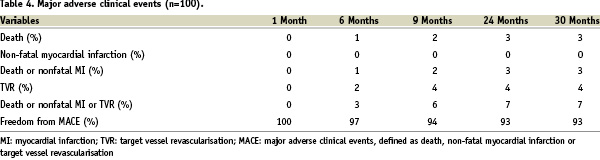
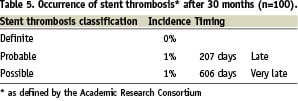
The event-free survival rate was 93% after 30 months follow-up. Four patients underwent TVR. Out of the three deaths, one patient died five months after the PCI of unknown causes; another died seven months after stent implantation due to progressive heart failure; the third patient died from a myocardial infarction after 24 months; all deaths were classified as cardiac according to Academic Research Consortium definitions (ARC)24. Using the ARC guidelines, the device orientated composite endpoint (cardiac death, myocardial infarction [not clearly attributable to non-target vessel], TLR), was reached in 6% and 7% of patients after one and two years respectively. There were no cases of definite stent thrombosis. The classification of stent thrombosis according to ARC criteria is shown in Table 5.
Discussion
The initial enthusiasm following the introduction of the first-generation of DES has since been somewhat cooled by long-term safety concerns, particularly regarding stent thrombosis and the clinical sequelae of myocardial infarction and death. The exact causes of these complications remains unclear, but both the drug and the permanent polymer coating have been implicated9-11,25. One potential modification of the next generation of DES is the use of a biodegradable polymer to reduce adverse interactions whilst allowing controlled drug release.
Polymer-based sirolimus-eluting stents have already been evaluated with mixed results: the CURA stent (Orbus Neich, Fort Lauderdale, FL, USA) consisting of a stainless steel stent with a PLA / PLGA / sirolimus matrix had angiographic restenosis rates of 22% and a late loss of 0.74 mm when used in patients presenting with acute myocardial infarction26. This compares poorly to the late loss with the first generation sirolimus-eluting stent (late loss of 0.17 in the SIRIUS trial1). The Excel stent (JW Medical Systems, China), a stainless steel stent with abluminal PLA / sirolimus performed much better, with a late loss of 0.07 mm27,28.
When comparing the angiographic efficacy for the Supralimus stent to the first generation sirolimus-eluting stent (Cypher: Cordis, Warren, NJ, USA), both the in-stent (late loss 0.09 mm for Supralimus vs. 0.17 mm for Cypher, angiographic restenosis 0% for Supralimus vs. 3.2% for Cypher) and in-segment (late loss 0.02 mm for Supralimus vs. 0.24 mm for Cypher, angiographic restenosis 1.7% for Supralimus vs. 8.9% for Cypher) results appear favourable1. The video-densitometric QCA results from the Supralimus stent (a decrease in in-stent minimum lumen area from 6.24±2.99 mm2 post-procedure to 5.63±2.67 mm2 after 6 months) also appear favourable when compared with the commercially available paclitaxel-eluting stent (Taxus: Boston Scientific, Natick, MA, USA) which had a corresponding decrease from 6.42±2.45 mm2 post-procedure to 4.51±2.42 m2 after six months29. In the Randomised Study with the Sirolimus-Coated Bx Velocity Balloon-Expandable Stent in the Treatment of Patients with de Novo Native Coronary Artery Lesions (RAVEL) study, an 18 mm stent was used to treat a mean lesion length of 9.56±3.33 mm2. In SERIES-1 we also ensured complete lesion coverage by using a mean stent length of 18.95±7.58 mm to treat a mean lesion length of 10.5±4.30 mm.
The clinical and safety profile of the Supralimus stent gives comparable results across all the clinical endpoints when scrutinised next to the two year meta-analysis of the pivotal US, European and Canadian randomised controlled trials of the Cypher stent (mortality 2.1%, TLR 5.7%, overall MACE 10.6% compared to 3%, 4% and 7% respectively for the Supralimus stent)30.
Beyond efficacy and safety, a further issue with DES is the high cost compared to bare metal stents, which may limit their use worldwide31,32. Hopefully the advent of drug-eluting stents developed and manufactured in South Asia will prove beneficial, since these stents are likely to be cheaper than those produced in the USA or Europe. The cost difference between Cypher and Supralimus is approximately 700 USD.
This study is a first-in-man single-centre clinical evaluation and therefore is limited by small patient numbers and the treatment of less complex patients than some of the available registries33. The long-term safety and efficacy of the biodegradable polymer-based Supralimus® stent needs to be established by future large-scale clinical trials. Nonetheless, the long term MACE-free survival and angiographic late loss appear to be comparable to other available DES.
