Abstract
Background: We studied the Nobori™ coronary stent coated with a bioabsorbable polymer and the anti-proliferative agent Biolimus A9 which may reduce neointimal formation.
Methods and results: Patients undergoing percutaneous coronary intervention for de novo lesions in up to two native coronary arteries, in 29 centres across Europe, Asia and Australia were randomly (2:1) assigned to receive the Biolimus A9 eluting stent Nobori™ (85 patients) or paclitaxel eluting stent Taxus® (35 patients). The two groups were well matched in baseline characteristics. The primary end point of non-inferiority for in-stent late loss of Nobori™ stent versus Taxus® stent, at 9 months, was reached with the values of 0.15±0.27 mm with Nobori™ stent and 0.32±0.33 mm with Taxus® stent (p=0.006). Neointimal volume obstruction was 2.2±6.0% and 8.9±9.2% for Nobori™ and Taxus® stent respectively (p=0.017). The rates of death, myocardial infarction and any target vessel revascularisation at 9 months were 0%, 4.7%, and 7.1% respectively for Nobori™ stent, and 0%, 8.6% and 14.3% respectively for Taxus® stent. Clinically-driven target lesion revascularisation rate was 0% for Nobori™ stent and 2.9% for Taxus® stent. Stent thrombosis rates at 9 months were 0% in both groups.
Conclusions: In this trial the Nobori™ Biolimus A9 eluting stent proved to be safe and effective in reducing neointimal proliferation. The long term safety remains to be confirmed during the extended follow-up period of 5 years.
Introduction
Coronary stents have revolutionised the field of interventional cardiology and stent implantation has become the standard of care in percutaneous coronary interventions. However, the long-term success of coronary stenting is hampered by in-stent restenosis which is caused by proliferation and migration of smooth muscle cells and production of extracellular matrix. Attempts at identifying an effective anti-restenosis therapy have been disappointing until recently when it was demonstrated that placement of coronary stents containing anti-proliferative agents reduced neointimal hyperplasia and the resulting restenosis1-7. However, long term safety concerns, mainly associated with delayed vessel healing, hypersensitivity on polymer carriers and late stent thrombosis, have prompted new developments in this field8-15. The main targets were more biocompatible polymers and drugs specifically developed for local application. The Nobori™ coronary stent system employs a bioabsorbable polymer (poly-lactic acid) from which Biolimus A9, an analogue of sirolimus, is eluted. The Biolimus A9 compound is expected to reduce neointimal proliferation while the polymer is expected to be absorbed within a few months. Since polymers have been implicated in some of the late adverse events related to DES13-15, the degradation of polymer is expected to improve long term safety of Nobori™ stent. The present study is the first clinical experience with the Nobori™ stent. It is a prospective, controlled, randomised (non-proportional 2:1), non-inferiority, two-arm trial of the Biolimus A9 eluting Nobori™ stent and the paclitaxel eluting stent Taxus®.
Methods
Patient population
Patients who were at least 18 years of age, with ischaemic heart disease due to de novo lesions in up to two native coronary arteries were considered for enrolment. Angiographic inclusion criteria were a reference vessel diameter of 2.5 mm to 3.5 mm and a lesion length > 5 mm and < 25 mm. Major clinical exclusion criteria were: left ventricular ejection fraction < 30%; myocardial infarction (MI) within the preceding 48 hours; intolerance to aspirin, heparin, clopidogrel bisulfate, ticlopidine, drugs similar to Biolimus A9 (sirolimus, tacrolimus, everolimus, zotarolimus), paclitaxel, contrast media, and stainless steel; platelet count <100,000 or >700,000 cells/mm3 or a WBC < 3,000 cells/mm3; serum creatinine level >2.0 mg/dL (or >150 µmol/L); current participation in other investigational trials; any coronary interventional procedure within 30 days before, or planned within 60 days after, the implantation of study stent; planned surgery within 6 months; stroke or transient ischaemic attack in the previous 3 months; gastrointestinal bleeding. Main angiographic exclusion criteria were: significant (> 50%) stenosis proximal or distal to the treated lesion; previous stenting anywhere in the territory of treated vessel; total occlusion (TIMI flow 0 and I); left main or ostial target lesion; severe calcification; evidence of thrombus or severe tortuosity.
The study patients were enrolled in 29 medical centres (listed in Appendix 1) from May to November 2005. The study was conducted according to the Declaration of Helsinki, ISO 14155-1 and -2, and respecting all country specific regulatory requirements. The protocol was reviewed and approved by the ethics committee of each participating medical institution. Prior to any test or procedure related to the trial, the benefits and the risks of the study were explained and written informed consent was obtained from each participating patient. An automated telephone randomisation system was used to assign eligible patients to treatment with Nobori™ or Taxus® stent in a 2:1 ratio before the stent implantation.
The Biolimus A9 eluting stent
The Nobori™ drug eluting stent system (Terumo Corporation, Tokyo, Japan) comprises three components: the stainless steel S-stent and its delivery catheter, a drug carrier, poly-lactic acid (PLA), and an anti-proliferative substance, Biolimus A9™ (Biosensors International). PLA has been used in a variety of medical applications and the final products of its degradation are carbon dioxide and water. The Nobori™ DES is coated only abluminally with a matrix containing Biolimus A9 and PLA (15.6 µg each per millimetre of stent length). The drug-polymer matrix is designed to release the drug in a two phase process. The first phase is a burst release immediately after stent implantation followed by the simultaneous process of sustained drug release and polymer degradation over several months. The coating design along with the lipophilicity of the drug, are expected to optimise drug distribution and to reduce its release into the peripheral circulation.
Biolimus A9 is an analogue of sirolimus, possessing immunosuppressive and anti-proliferative activities. Biolimus A9 offers promise in preventing restenosis due to its expected ability to interrupt cell migration and proliferation by arresting cell cycle in the late G1 phase. The potency of locally delivered Biolimus A9 to prevent neointimal proliferation has been confirmed in the STEALTH study5 which compared BioMatrix® Biolimus A9 eluting stent to bare metal stent.
In this current study the polymer based paclitaxel eluting stent Taxus® (Boston Scientific Corporation) was used as comparator. Safety and efficacy of the polymer based paclitaxel eluting stent has been confirmed in numerous studies3,4,16.
Coronary stent procedure
At least 100 mg of aspirin was recommended daily to all patients before the procedure and indefinitely thereafter. A loading dose of 300 mg of clopidogrel was recommended if administered more than 6 hours before the procedure and 600 mg if administered periprocedural. Clopidogrel was further recommended at 75 mg/day for at least 6 months to all patients receiving stents. During the procedure intravenous heparin boluses were administered per standard hospital practice and intravenous glycoprotein IIb/IIIa inhibitor use was at the physician’s discretion.
Nobori™ stents were available in lengths of 8, 14, 18, 24 and 28 mm and in diameters of 2.5, 3.0 and 3.5 mm. Taxus® stents were available in lengths of 8, 12, 16, 20, 24 and 28 mm and in diameters of 2.5, 3.0 and 3.5 mm. After mandatory predilatation, an appropriately sized stent (2-4 mm longer than the lesion, with a stent to distal reference vessel-diameter ratio of 1-1.1:1) was implanted. Additional study stents were permitted for edge dissection or otherwise suboptimal results. Post-dilatation was allowed at the operator’s discretion, and use of a balloon shorter than the stent was recommended. Any second lesion in a second coronary vessel had to be treated in the same manner.
Pre-procedural and post-procedural ECGs were obtained and cardiac enzymes were measured at baseline, 8-12 hours and 18-24 hours after the procedure or at discharge whichever came first.
Patient follow-up
All surviving patients were scheduled for a control angiography at 9 months ± 30 days along with intra-vascular ultrasound (IVUS) in the pre-specified sites which enrolled 49% of the patients. Clinical follow-up was scheduled at 30 days, 4, 9 and 12 months and yearly thereafter up to 5 years. The clinical follow-up included assessment of angina status, monitoring use of cardio-active and anti-thrombotic drugs, hospitalisations, occurrence of major adverse cardiac events, and any invasive and non-invasive diagnostic test or interventional treatment that occurred since the previous contact.
Study management
A Data Safety and Monitoring Board was responsible for the review of data and identification of potential safety issues. The members of this Board were not affiliated with the study sponsor and were not participating in this trial.
An independent clinical event committee reviewed and adjudicated all major adverse cardiac events. The committee was composed of interventional cardiologists, not associated with the sponsor.
The study was managed by Cardialysis (Rotterdam, The Netherlands), an independent contract research organisation (CRO) which was also responsible for monitoring, data management and analysis.
Endpoints and definitions
The primary end point was angiographic in-stent late-loss at 9 months post-procedure defined as the difference between the post-procedure minimal lumen diameter (MLD) and the follow-up angiography MLD. Secondary endpoints were MACE, defined as: cardiac death; MI (Q wave and non-Q wave); emergent cardiac bypass surgery; target vessel revascularisation (TVR) at 30 days, 4, 9, and 12 months, and yearly up to 5 years; angiographic in-stent and in-segment binary restenosis rate (defined as > 50% diameter stenosis); in-stent, in-segment, proximal, and distal MLD at 9 months post-procedure; target lesion revascularisation (TLR) at 9 months post-procedure; stent thrombosis at 30 days and nine months; neointimal hyperplasia volume at 9 months post-procedure as measured by IVUS; lesion success defined as the attainment of < 30% residual stenosis by visual assessment and/or < 50% by QCA using any percutaneous method; device success defined as achievement of a final diameter stenosis of < 50% by QCA, and/or < 30% by visual assessment, using the assigned device only; procedure success defined as achievement of a final diameter stenosis of < 30% by visual assessment and/or < 50% by QCA, using any percutaneous method, without the occurrence of death, Q wave or non-Q wave MI, or repeat revascularisation of the target lesion during the hospital stay. MI was defined either as the development of pathological Q-waves in at least two contiguous leads with or without elevated cardiac enzymes or, in the absence of pathological Q-waves, as an elevation in creatinine kinase levels to greater than twice the upper limit of normal in the presence of an elevated level of CK-MB fraction. TLR was defined as any clinically-driven repeat percutaneous intervention of the stented lesion including 5 mm proximal and distal from the edge of the stent, or bypass surgery of the target vessel that was performed for a clinical indication and was due to restenosis or closure of the target lesion. TLR was considered clinically-driven if prompted by a positive functional study, by ischaemic ECG changes at rest in a distribution consistent with the target vessel, or by ischaemic symptoms with an in-lesion diameter stenosis > 50% by QCA or if lesion diameter stenosis was more than 70% at follow-up. Target vessel revascularisation was defined as revascularisation (clinically- or not clinically-driven) of any segment of the index coronary artery, which was in physical contact with any component (guiding catheter, guidewire, balloon catheter, etc.) of the angioplasty hardware during the initial procedure. Stent thrombosis was defined as confirmed thrombus within the stented vessel at the time of the clinically-driven angiographic restudy for documented ischaemia (chest pain and ECG changes), representing abrupt or sub-abrupt closure. Any death (without other obvious cause), or any Q-wave myocardial infarction in the territory of the stented segment within the first 30 days were considered a surrogate for stent thrombosis when angiography was not available. It was classified as acute if it occurred within the first 24 hours, sub-acute up to 30 days, and late after 30 days. Late thrombosis was defined as MI attributable to the target vessel with angiographic documentation of thrombus or total occlusion at the target site more than 30 days after the index procedure in the absence of an interim target vessel revascularisation.
Quantitative coronary angiography and IVUS evaluation
An independent angiographic core laboratory (Cardialysis, Rotterdam, The Netherlands) analysed all pre-, peri- and post-procedural angiographic images using edge detection technique (CAAS II, Pie Medical, Maastricht, the Netherlands). In each patient all pre-, post-procedural and follow-up angiographic images were analysed. The following parameters were computed: reference vessel diameter (RVD), minimal lumen diameter (MLD) and percent diameter stenosis. Binary restenosis (BR) was defined as diameter stenosis of equal or more than 50% at follow-up. Late loss (LL) was defined as the difference between MLD post-procedure and MLD at follow-up.
Post-procedure and follow-up stented vessel segments were examined with IVUS using automated pullback at 0.5 mm per second. A coronary segment beginning 5 mm distal to and extending 5 mm proximal to the stented segment was also examined. A computer-based contour detection program (Curad B.V., Wijk bij Duurstede, The Netherlands) was used for automated 3-D reconstruction of the stented and the peri-stent segments. The lumen, stent boundaries and external elastic membrane were detected using a minimum cost algorithm17. The stent volume (SV) and lumen volume (LV) were calculated according to Simpson’s rule18. The in-stent neointimal volume was calculated as “SV minus LV”. The percent obstruction of the stent volume was calculated as in-stent neointimal volume divided by stent volume and multiplied by 100. Feasibility, reproducibility and inter- and intra-observer variability of this system have been validated in vitro and in vivo17.
Statistical analysis
The primary analysis of this study has taken into account that this was a randomised, 2-arm, non-inferiority trial, using the primary end point in-stent late loss at 9 months as a target. A 0.20 mm difference in late loss was considered to be the minimum clinically important difference. Therefore, the study was designed to prove that the upper confidence limit of the late loss at 9 months for the Nobori™ stent was, on average, not more than 0.20 mm higher than the comparator late loss. The power of the trial was set at 90% (β=10%). The one-sided type-I error (α) was determined to be 2.5%. The assumptions used for power calculations were 0.26±0.43 mm late loss at 6-months recorded in STEALTH study5 for the Biolimus A9-eluting stent and 0.39±0.50 mm late loss at 9-months recorded in TAXUS-IV clinical trial16. After consideration of historical drug eluting stent outcomes, the result of Biolimus A9- eluting stent was extrapolated to an estimated 0.39±0.50 mm late loss at 9 months. Applying these assumptions and the unbalanced (2:1) study design, a sample size of 300 lesions was estimated for the trial to prove non-inferiority. The total sample size was increased to 360 patients to accommodate for ~80% compliance with 9-months angiographic follow-up taking into account that up to two lesions were allowed in the study. However, after 120 patients were enrolled in the study (85 patients/95 lesions in Nobori™ arm and 35 patients/42 lesions in Taxus® arm), the sponsor, in consultation with the data safety and monitoring board and the steering committee, decided to temporarily suspend the study and split it into two phases due to the need to improve the retention force for the Nobori™ stent (three stent dislodgments were observed) and because of replacement of Taxus® Express™ with Taxus® Liberté™ in the European market. The primary end point, in-stent late loss, in this phase of the study was therefore calculated by reduced power.
The primary end point and all trial end points were analysed on the intent-to-treat population. Clinical events including death, MI, and revascularisation are reported on a per patient basis. For patients with multiple lesions, a failure of any lesion was counted toward the composite event rate. For continuous variables, differences between the treatment groups were examined by analysis of variance, while Fisher’s exact test was used for categorical variables. All statistical analyses were performed using SAS statistical software, version 8 (SAS Institute Inc, Carry, NC, USA).
Results
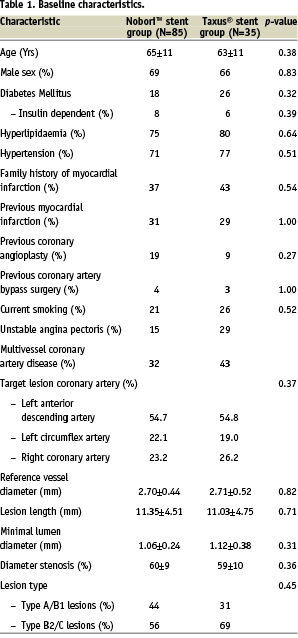
Between May and November 2005, 120 patients were randomly assigned in a 2:1 ratio to receive either the Nobori™ Biolimus A9 – eluting stent or the Taxus® paclitaxel – eluting stent. This cohort constitutes the first phase of the trial. There were 85 patients with 95 lesions assigned to treatment with Nobori™ stents and 35 patients with 42 lesions assigned to Taxus® stent. The baseline characteristics of the two groups were well matched (Table 1). The age of patients and the rate of diabetes mellitus were representative of current PCI practice and of the areas of patient’s enrolment.
Baseline lesion and procedural characteristics
The lesion characteristics and treated vessel distribution were similar between the groups (Table 1). More than half (55%) of the target lesions were located in the left anterior descending coronary artery, while other lesions were evenly distributed between the left circumflex and the right coronary artery.
All lesions were predilated prior to stent deployment. There were 2 (2.1%) dissections in the Nobori™ arm and 7 (16.7%) in the Taxus® arm. Mean deployment pressures were similar with 13.8 and 14.0 atmospheres in Nobori™ and Taxus® arms respectively. Both arms had similar success rates for lesion (100% both groups), device (97.9% and 97.6% for Nobori and Taxus respectively) and procedure (95.2% for Nobori™ and 88.2% for Taxus® stent). Bifurcation stenting requiring double guidewire was similar in the two groups (7% in Taxus® and 8% for Nobori™ arm). Use of platelet glycoprotein IIb/IIIa inhibitors was very low in both groups (less than 3%). In the Nobori™ stent group there were 3 stent dislodgments which did not result in any adverse clinical event. Two stents were successfully implanted into the target lesion despite partial dislodgment, while the third stent dislodged from the balloon before reaching the coronary artery and was found in the guiding catheter.
Quantitative coronary angiography findings
Angiography at 9-months was completed in 101 (84%) patients with 115 (84%) lesions. Pre-procedural angiographic lesion characteristics were similar in the two groups (Table 2). At 9 months mean in-stent late loss, the primary end point of the study, was significantly lower for the Nobori™ stent arm compared to the Taxus® arm, 0.15±0.27 mm versus 0.32±0.33 mm (p<0.006). The upper limit of the confidence interval of the difference in late loss was -0.07 mm suggesting that the Nobori™ stent is non-inferior versus the Taxus® stent. The cumulative frequency distribution of late loss for the two studied stents is shown in Figure 1. Other relevant QCA parameters such as MLD, absolute gain, diameter stenosis after procedure and at 9 months follow-up were similar in both groups (Table 2).
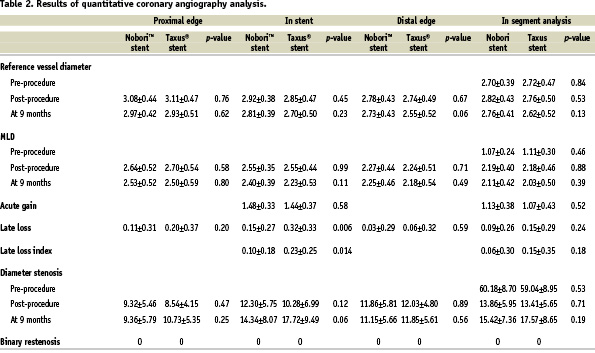
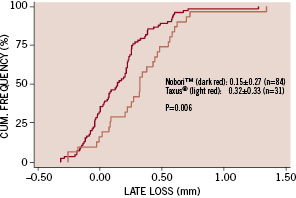
Figure 1. Cumulative frequency of in-stent late loss at 9 months follow-up.
Intravascular ultrasound evaluation
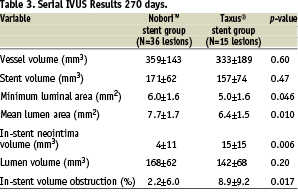
IVUS was also used to assess vessel, stent, and lumen volume, plaque volume and in-stent volume obstruction after stent implantation and at 9 months in a total of 51 lesions (37%). The vessel volume within the stented segment, stent volume and lumen volume were not different in the Nobori™ and Taxus® groups either at baseline or at follow-up. However, at 9 months follow-up, the minimum and mean lumen area, mean plaque area and in-stent volume obstruction were all significantly different favouring the Nobori™ stent (Table 3).
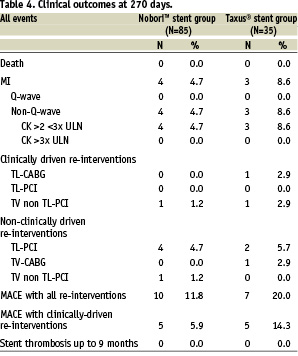
Clinical outcomes
In hospital
During the index hospitalisation, 4 (4.7%) patients treated with the Nobori™ stent suffered non-Q-wave MI with mild elevation of cardiac enzymes. The values of creatine kinase enzyme did not exceed 3 times the upper normal level in any of the patients. During the same study period, 3 (8.6%) patients treated with the Taxus® stent suffered non-Q-wave MI, and 1 (2.9%) patient underwent emergent CABG procedure. The overall rate of in-hospital major adverse cardiac events was 4.7% for the Biolimus A9 eluting stent group and 11.4% for the Paclitaxel-eluting stent group.
During clinical follow-up
Clinical follow-up at 30 days and 4 months was completed for 100% and at 9 months for 95.8% of the patients. No further adverse cardiac events occurred between hospital discharge and 30 days. Between 1 and 4 months the only reported major adverse cardiac event was one non clinically-driven CABG procedure in the Taxus® group.
At 9 months follow-up there were no deaths, no new myocardial infarctions, or clinically driven target lesion revascularisations. Non clinically-driven target lesion revascularisation rate was 4.7% (n=4) in the Nobori™ stent group and 5.7% (n=2) in the Taxus® group. All TLRs were performed in lesions with less than 50% diameter stenosis. The incidence of target vessel, non-target lesion revascularisation was 2.4% (n=2) in the Nobori™ arm and 2.9% (n=1) in the Taxus® arm. One target vessel revascularisation in the Nobori™ arm was non clinically-driven. Cumulative MACE rate at 9-months follow-up, including all target vessel revascularisations, was 11.8% for the Nobori™ stent and 20.0% for the Taxus® stent. The cumulative MACE rate considering only clinically-driven revascularisations was 5.9% in the Nobori™ arm and 14.3% in the Taxus® arm. No patients in this study suffered an acute, sub-acute nor late stent thrombosis.
Compliance with dual antiplatelet therapy
The overall mean (SD) duration of dual antiplatelet therapy was 236 (45) days in the Nobori™ arm versus 243 (45) days in Taxus® stent group. This difference was not statistically significant. At 6 months 100% patients in Nobori™ group and 90.6% of the patients in Taxus® group were treated with doses of clopidogrel or ticlopidine according to the protocol-mandated guidelines (p=0.021).
Discussion
This prospective, randomised, multicentre study was the first clinical experience with the Nobori™ Biolimus-A9 – eluting coronary stent. The Nobori 1 trial has met its primary end-point, the non-inferiority of in-stent late loss of the Nobori™ stent versus the Taxus® stent. Both studied stents showed a good safety profile without deaths, Q-Wave myocardial infarction and stent thrombosis up to 9 months.
Despite the low number of patients and the excellent anti-proliferative performance of the paclitaxel eluting stent in this trial, the promising efficacy of locally delivered Biolimus A9 is supported by significant and concordant improvements in the QCA and IVUS parameters.
At 9 months angiographic and IVUS follow-up significant differences were found in in-stent late loss, in-stent late loss index, minimum and mean lumen area, mean plaque area and in-stent volume obstruction, all favouring the Biolimus A9-eluting stent. These observations indicate a greater degree of suppression of neointimal hyperplasia. Although Nobori 1 clinical trial has not been powered to prove superiority of Nobori DES over Taxus DES in terms of in-stent late loss, the magnitude of difference indicates that with larger number of patients this assumption could be confirmed. However, due to the predominantly simple lesions treated and the small number of patients enrolled in this study, significant differences in several continuous variables, most importantly in-stent late loss, did not translate into significant differences in in-lesion binary restenosis or in target lesion revascularisation.
Although both Biolimus A9 and paclitaxel inhibit neointimal proliferation, they don’t share the same mechanism of action. In addition to the drug specificities, the coating materials and coating design may result in different drug release kinetics and uptake of the drug into the arterial wall. It remains unclear which component of the two studied stent systems contributed to the different efficacy in the neointimal volume suppression.
Overall MACE rate in this trial is probably exaggerated by the inclusion of all target vessel revascularisations whether clinically driven or not, and by a slightly higher rate of procedural non-Q-Wave myocardial infarctions. Morice et al in the REALITY trial19, also allowing treatment of up to 2 lesions, found combined rate of Q-wave and non-Q-wave MI during index hospitalisation of 4.2% and 4.6% for sirolimus-eluting and paclitaxel-eluting stents, respectively. These results are similar to procedural MI rate reported for Nobori™ stent, whereas small number of patients treated with Taxus® stent had probably exaggerated relative ratio of MI in this study group. The clinical significance of isolated cardiac enzyme elevation following otherwise successful procedure remains controversial. In the analysis of patients enrolled in 6 major clinical trials of native coronary artery stenting (6,186 patients) Jeremias et al20, found that procedural non-Q-wave MI, particularly with moderate enzyme increase, was not associated with an increase of 1 year mortality rates after adjustment to unsuccessful procedure. The rate of procedural myocardial infarctions is often subject to the markers used, cut-off values and measurement time points. In a recently published paper Wiseth et al21, called for a separate ICD code for procedural MI in order to clearly separate it from spontaneous MI.
The rather high rate of non clinically-driven target lesion revascularisations corroborates the magnitude of the occulo-stenotic reflex22,23 associated with angiographic evaluation at follow-up.
Comparison of Biolimus-A9- eluting stent with other drug eluting stents
The neointimal volume suppression recorded for the Biolimus A9 eluting stent resembles the suppression previously reported for sirolimus1,2,19 and everolimus6 eluting stents, while it appears superior to zotarolimus7 and paclitaxel3,4,19 eluting stents. Another Biolimus A9 eluting stent studied in the STEALTH trial showed similar efficacy5. Due to the discernible differences in the definitions of adverse events across the trials, a comparison of overall MACE rate appears inappropriate. However, the absence of angiographic restenosis and clinically-driven target lesion revascularisation up to 9 months, along with no incidence of death, post procedural MI and stent thrombosis indicate a good safety profile of the Nobori™ stent.
Study limitations
Although the present study was a prospective, multicentre, randomised, controlled trial, several limitations are noteworthy. The reduced number of enrolled patients versus the original study design with a great impact on power of the analyses is the main limitation of this trial. The second phase of this trial has finished enrolment and the results of that part of this trial may confirm these initial findings. Selection of an angiographic primary endpoint rather than clinical could be considered as a limitation of the study, however, in the early stages of stent development late loss as an intuitive measure of neointimal hyperplasia, the pathological target of drug-eluting stents, appears justified. Moreover, in-stent late loss has proven to be a good predictor of TLR24. The safety and efficacy of the Biolimus A9 eluting Nobori™ stent in our study population cannot be generalised to patients and types of lesions that were excluded from randomisation. Therefore larger studies with less restrictive eligibility criteria are needed to substantiate the benefits of the biodegradable polymer and sustained Biolimus A9 release for patients with more complex lesions and higher risk patients. Also this study was not powered to detect clinical non-inferiority of Nobori stent versus Taxus® stent and rare events like stent thrombosis with sufficient confidence intervals.
Conclusion
In this first head-to-head comparison to an approved DES, the Nobori™ Biolimus A9-eluting stent showed its non-inferiority with higher reduction of neointimal hyperplasia, translated into absence of angiographic restenosis and clinically-driven target lesion revascularisation. In addition to its satisfactory anti-proliferative performance, the Nobori™ stent appeared safe in the studied population with no death, post-procedural myocardial infarction and stent thrombosis up to 9 months follow-up. Further studies enrolling larger number and higher risk patients are either ongoing or planned and their results are anticipated to confirm these initial findings.
Acknowledgements
The authors would like to express great appreciation to all patients who agreed to take part in this study and to acknowledge the invaluable assistance of Danny Detiege from Terumo Europe and Monique Schuijer from Cardialysis for managing the trial.
Appendix 1
Nobori 1 Study Organisation
Sponsor: Terumo Europe N.V. Leuven, Belgium
Nobori 1 Principal Investigator: Bernard Chevalier, Centre Cardiologique du Nord, Saint-Denis, France
The Nobori 1 Steering Committee: Bernard Chevalier, Centre Cardiologique du Nord, Saint-Denis, France; Patrick Serruys, Erasmus MC Thoraxcentrum, Rotterdam, The Netherlands; Eulogio Garcia, Gregorio Marañón, Madrid, Spain; Sigmund Silber, Kardiologische Gemeinschaftspraxis und HKL, München, Germany; Harry Suryapranata, Isala Clinic, Zwolle, The Netherlands; Hirofumi Nagai, Terumo Corporation, Tokyo, Japan; Monique Schuijer, Cardialysis, Rotterdam, The Netherlands; Dragica Paunovic, Terumo Europe, Leuven, Belgium.
Clinical Event Commitee: Gian Battista Danzi, Ospedale Maggiore Policlinico, Milan, Italy; Philippe Urban, Hopital de la Tour, Geneva, Switzerland; Bernhard Reimers, Ospedale di Mirano, Mirano, Italy; Claude Hanet, Cliniques Universitaire St.-Luc, Brussels, Belgium.
The Nobori 1 Clinical Investigators
Australia: Stephen Worthley, Royal Adelaide Hospital, Adelaide, South Australia; Ian Meredith, Monash Medical Centre, Clayton, Victoria; Belgium: William Wijns, Onze-Lieve-Vrouwziekenhuis, Aalst; Marc Castadot, Clinique St-Jean, Brussels; Victor Legrand, Centre Hospitalier Universitaire de Liège, Liège; Denmark: Leif Thuesen, Skejby Sygehus, Aarhus; France: Bernard Chevalier, Centre Cardiologique du Nord, Paris; Martial Hamon, Centre Hospitalier Universitaire Caen, Caen; Marie-Claude Morice, Institut Jacques Cartier ICPS, Massy; Didier Carrie, Centre Hospitalier Universitaire Rangueil, Toulouse; Philippe Garot, Hôpital Claude Gallien, Quincy; Jean Marco, Clinique Pasteur, Toulouse; Emmanuel Teiger, Hôpital Henri Mondor, Créteil; Germany: Karl Hauptmann, Krankenhaus der Barmherzigen Brüder, Trier; Sigmund Silber, Kardiologische Gemeinschaftspraxis und HKL, München; Gerhard Schuler, Universitätsklinikum Leipzig/ Herzzentrum, Leipzig; Nicolaus Reifart, Kardiologische Praxis Professor Reifart & Partner, Bad Soden; Christian Hamm, Kerckhoff Klinik GmbH, Bad Nauheim; Bernd Nowak, Bethanien Krankenhaus, CCB, Frankfurt; Thomas Schiele, Klinikum Innenstadt der LMU, München; Korea: Seung-Jung Park, Asan Medical Center, Seoul; The Netherlands: Patrick Serruys, Erasmus MC Thoraxcentrum, Rotterdam; Harry Suryapranata, Isala Clinic, Zwolle; JJ Bonnier, Catharina Ziekenhuis, Eindhoven; Spain: Eulogio Garcia, Gregorio Marañón, Madrid; Antoni Serra, Hospital del Mar, Barcelona; United Kingdom: F Fath-Ordoubadi, Royal Infirmary, Manchester; Martyn Thomas, Kings’ College Hospital, London; David Hildick-Smith, Royal Sussex County Hospital, Brighton.
