Abstract
Background: SIMPLE II was a multi-centre, prospective registry study aimed at investigating the safety and efficacy of the Infinnium™ (Sahajanand Medical Technologies Pvt. Ltd, India) paclitaxel-eluting stent for the treatment of single de novo lesions in the native coronary arteries.
Methods: One hundred and three patients with symptomatic coronary artery disease were treated for single de novo native coronary artery lesions using the Infinnium™ stent (paclitaxel concentration 1.4 mcg/mm2 released over 48 days) in a multi-centre, prospective study performed on 3 continents (Asia, Europe and South America). The primary safety endpoint was major adverse cardiac events at 30 days (MACE 30d) and efficacy was assessed by in-stent binary restenosis as measured by quantitative coronary angiography (QCA) at six-month follow-up. A clinical follow-up was scheduled at nine months.
Results: The mean patient age was 58.5 years; 70.9% were males; 43.7% had unstable angina and 38.8% previous myocardial infarction. Risk factors included hypertension in 62.1%, hypercholesterolemia in 52.4%, current smoking in 32.0% and diabetes in 28.2%. Stent implantation was successful in all patients, with more than one stent being implanted in 9 patients (8.7%). Hierarchical MACE 30d was 2.9%. At nine months, 101 patients had clinical follow-up (1 patient had died and 1 refused). There was one death (1.0%), one Q-wave myocardial infarction (Q MI) (1.0%), three non-Q MIs (2.9%), one clinically-driven target lesion Coronary Artery Bypass Grafting (CABG) (1.0%), and one clinically-driven target lesion repeat percutaneous coronary intervention (re-PCI) (1.0%). The overall event-free rate at nine months was 93.2%. QCA revealed in-stent and in-segment late loss of 0.38±0.49 mm and 0.18±0.46 mm, resulting in binary restenosis rates of 7.3% and 8.3%, respectively. There was one case of late stent thrombosis in the patient experiencing the Q MI and subsequent re-PCI.
Conclusions: The Infinnium™ paclitaxel-eluting stent appears to be safe and efficacious for the treatment of single de novo lesions in coronary arteries in a patient population with symptomatic coronary artery disease (CAD).
Introduction
Although the immediate success and safety of coronary stenting has dramatically increased, in-stent restenosis has persisted as a limitation hampering the medium-term efficacy of coronary stenting.1,2 The elucidation of the molecular and cellular mechanisms of inflammation and cellular proliferation in vascular injury and repair powered the development of site-specific and controlled release of therapeutic agents through the drug-eluting stent (DES) technology.3,4,5,6 The cell cycle is a common hub of the different phases of the restenosis process.7 Antiproliferative agents, such as sirolimus and paclitaxel have been coupled with polymers that elute or slowly release these inhibitors from the stent surface. Variations in pharmacological mechanisms and stent-coating technologies elicited different vascular reactions.8,9,10,11,12,13,14
Paclitaxel (Taxol; Bristol-Myers Squibb), a natural diterpenoid compound extracted from the Pacific yew tree Taxus brevifolia, has diverse mechanisms of action, including microtubule stabilisation, arrest of cell mitosis, retardation of cell migration and immunomodulation.15,16,17,18,19,20,21 Paclitaxel is also highly lipophilic and poorly soluble in aqueous solution, making it an excellent candidate for sustained delivery from stents and prolonged deposition in atherosclerotic vessels. Stents coated with a polymer (lactide-co--caprolactone) to control the release of the drug have already proved effective in reducing late loss and restenosis in a series of clinical trials, the TAXUS studies.9,22,23
Many different platforms that use polymer coating or surface modifications to adhere paclitaxel onto the stents have been utilised over the past two years. The biodegradable-polymer-based, paclitaxel-eluting Infinnium™ stent (Sahajanand Medical Technologies Pvt. Ltd, India) is the first indigenously designed and evaluated DES from Asia.
The purpose of the present study was to assess the safety and efficacy of the Infinnium™ stent in single de novo native coronary artery lesions.
Methods
Selection of patients
The study was a non-randomised registry trial performed at 8 medical centres (with a maximum patient inclusion of 25 patients per site) on 3 continents (listed in the Appendix). This protocol was approved by the hospital ethics committees and is in accordance with the Declaration of Helsinki. All patients gave written informed consent.
Patients were eligible for the study if they were at least 18 years old, were not of child-bearing potential, and had received a diagnosis of symptomatic ischaemic heart disease: stable or unstable angina (CCS class 1-4, Braunwald class IB, IC, IIB, IIC, IIIB, IIIC)24 and/or objective evidence of myocardial ischaemia.
Additional eligibility criteria were the presence of a single primary target lesion in a native coronary artery that was 2.5 to 3.5 mm in diameter and that could be covered by one single study stent; if the coronary artery lesion was <10 mm (visual estimate) it had to be covered with a 19 mm stent and if it was >10 mm and <15 mm, it had to be covered with a 23 mm stent aiming for a lesion/stent ratio of at least 1.6. The luminal diameter of the lesion had to have a stenosis of 51-99%, as estimated visually, and a flow rate grade of 1 or more according to the classification of the thrombolysis in myocardial infarction (TIMI) trial.
Patients were not eligible for enrolment if they had any significant medical condition which could interfere with the patient’s optimal participation in the study, a life expectancy of less than 12 months, a Q-wave or non-Q-wave myocardial infarction within 72 hours preceding the index procedure (unless the CK and CK-MB enzymes were less than twice the Upper Normal Limit), other revascularisation procedures within the previous 6 months or prior stenting within 5mm of the target lesion, an unprotected left main coronary artery stenosis (>50%), an ostial lesion, a calcified lesion that could not be completely dilated before stenting, proximal tortuosity, angiographic evidence of thrombus within the target lesion, pre-treatment with devices other than balloon angioplasty, total occlusion, bifurcations (potentially requiring stenting of the side branch); another stenosis of more than 50% proximal or distal to the target lesion or in an other vessel, poor distal run-off, a left ventricular ejection fraction <30%, or an intolerance to aspirin, clopidogrel, ticlopidine, heparin, nickel, or contrast material. Patients were not allowed to be participating in another investigational drug or device study.
The Infinnium™ Paclitaxel-Eluting coronary stent
The active ingredient in the Infinnium™ stent is Paclitaxel. The paclitaxel concentration loaded on each stent was maintained to 1.4 µg/mm2. The drug was applied to the surface of a stainless steel (slotted tube design), balloon-expandable stent (Matrix, Sahajanand Medical Technologies Pvt.Ltd.) using biodegradable polymers (Poly L-Lactide, 50/50 Poly DL-Lactide-co-Glycolide, 75/25 Poly L-Lactide-co-Caprolactone and Polyvinyl Pyrrolidone) in multiple layers. The drug is coated in 3 different layers of combination of drug and polymer. Each layer has a different release profile. The cumulative release of drug from the polymer is at 48 days after implantation (see Figure 1).
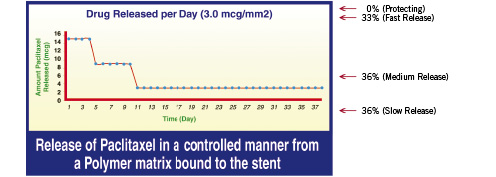
Figure 1. The Infinnium™ paclitaxel-eluting coronary stent.
Study procedures
Lesions were treated with the use of contemporary interventional techniques. Predilatation was advised but direct stenting was also allowed. The investigators used similar materials and techniques throughout the study to maintain consistency and standardisation of care.
The investigator determined the appropriate diameter (2.5, 2.75, 3.0 and 3.5 mm) of the Infinnium™ stent to be implanted aiming for a stent/vessel ratio of 1.1:1 prior to stent placement (using the nominal pressure). Two different stent lengths were recommended aiming for a lesion/stent ratio of at least 1.6. If the lesion was <10 mm, it had to be covered with a 19 mm stent, and if it was >10 mm and <15 mm, it had to be covered with a 23 mm stent.
After the stent was implanted, further dilation was performed as necessary to ensure that the residual stenosis (by on-line QCA) was <20% and the mean reference diameter of the stent was >1.1 mm with respect to the reference diameter of the adjacent segments, with a TIMI grade III flow rate. In case of an edge dissection, an additional 11 or 16 mm Infinnium™ stent could be deployed adjacent to the first stent in order to cover the dissection.
Intravenous boluses of heparin were administered to maintain an activated clotting time that exceeded 250 seconds during the procedure and were discontinued immediately following the index PCI procedure in order to allow for sheath removal. Treatment with aspirin, at a dose of 160-500 mg (when the patient was not already on aspirin), was begun 12 hours before the procedure and continued indefinitely. A loading dose of (at least) 300 mg of clopidogrel was administered optimally 48 hours before the index procedure with a minimum of 4 hours, followed by (at least) 75 mg daily for 6 months. Alternatively, treatment with ticlopidine, at a dose of 250 mg twice daily, was begun one day before the procedure and continued for 6 months. The administration of a glycoprotein IIb/IIIa inhibitor was optional and left to the investigator’s discretion according to clinical practice.
Follow-up
Patients were evaluated at 30 days (±7 days) and at 6 and 12 months (±30 days). They were asked specific questions about the interim development of angina, according to the Canadian Cardiovascular Society classification of stable angina25 and the Braunwald classification of unstable angina.26 The patients were also monitored for major cardiac events and for the need for additional revascularisation of the index target lesion. An electrocardiogram was obtained at each visit, and an angiographic study was performed at a mean (±SD) of 183±19.3 days. A 6-month post-procedure follow-up angiography – independent of the clinical status – was requested per protocol. Other studies and tests were performed at the discretion of the investigators at the participating centres.
The decision to perform further revascularisation of the target lesion or vessel after the six-month angiographic study was based on clinical justification (see definition of clinically driven target lesion revascularisation under “Study endpoints”).
Quantitative Coronary Angiographic (QCA) evaluation
Coronary angiograms were obtained in multiple views after the intracoronary injection of nitrates. Quantitative analyses of all angiographic data before, during, and after the procedure were performed by an independent core laboratory (Cardialysis, Rotterdam, The Netherlands) with the use of edge detection techniques (CASS II, Pie Medical).27 Visual assessments included TIMI flow, calcification, thrombus, lesion length, AHA/ACC classification, dissection grade and aneurysm. Interpolated reference diameter, minimal luminal diameter, and diameter stenosis were measured before dilation, at the end of the procedure, and at six months. Restenosis was defined as stenosis of >50% of the luminal diameter and was classified as in-stent if inside the stent or in-segment if located within the stented segment plus the 5-mm segments distal or proximal to the stent margins.28 Late luminal loss was calculated as the difference between the minimal luminal diameter immediately after the procedure and the diameter at six months.
Study endpoints
The primary goal included a reduction in the in-stent binary restenosis rate (luminal narrowing of ≥50%) at 6 months follow-up, as determined by quantitative angiography (QCA). Secondary goals included angiographic and procedural success, and a number of in-stent and in-segment (stent +5 mm proximal and 5 mm distal) vessel parameters derived by off-line QCA: acute gain, the minimal luminal diameter (MLD), the percentage of stenosis of the luminal diameter (% DS) and the mean lumen diameter.
The primary goal in this study also included a reduction of the composite of major adverse cardiac events (MACE) until 30 days, defined as cardiac death, Q-wave or non-Q-wave myocardial infarction, coronary artery bypass grafting (CABG) and clinically driven target lesion revascularisation (TLR). The secondary clinical endpoints were MACE until 9 months, device-related serious adverse events (DSAEs) until 9 months, and angiographic stent thrombosis (subacute: until 30 days, and late: until 9 months).
A non-Q-wave myocardial infarction was defined by an increase in the creatine kinase level to more than twice the upper limit of the normal range, accompanied by an increased level of creatine kinase MB, in the absence of new Q waves on the surface electrocardiogram. Angiographic success was defined as a successful delivery and deployment of the study stent – without the use of a device outside the study treatment strategy – and a final residual stenosis of <20% (by off-line QCA) with TIMI 3 post-procedure. Procedural success was defined as angiographic success in the absence of MACE during hospital stay. Target lesion revascularisation was defined as a repeat intervention (surgical or percutaneous) to treat a luminal stenosis within the stent or in the 5-mm distal or proximal segments adjacent to the stent. A TLR/TVR was considered clinically driven (retrospective adjudication by the clinical event committee) if the diameter stenosis at the time of angiography was >50% and at least one of the following criteria was present: a history of recurrent angina pectoris presumably related to the target vessel, objective signs of ischaemia at rest (ECG changes) or during exercise test (or equivalent) presumably related to the target vessel, abnormal results of any invasive functional diagnostic test (e.g. Doppler flow velocity reserve, fractional flow reserve). A TLR/TVR with a diameter stenosis >70% in the absence of the above- mentioned ischaemic signs or symptoms was also considered clinically driven. Thrombotic stent occlusion was angiographically documented as a complete occlusion (TIMI flow 0 or 1) or a flow-limiting thrombus (TIMI flow 1 or 2) of a previously successfully treated artery. The endpoints were adjudicated by an independent clinical event committee (CEC). In addition, an independent data and safety monitoring board (DSMB) that was not affiliated with the study sponsor reviewed the event data to identify any safety issues related to the study (see Appendix 1 for members of the CEC and DSMB).
Statistical analysis
This is an observational, prospective, non-randomised registry. No formal power calculation was performed. The primary analysis was carried out according to the intention-to-treat principle.
Descriptive statistics was performed for all relevant variables. Count variables were summarised by the count and the percentage. Various continuous variables were summarised by the mean, standard deviation, minimum and maximum. The event variables, such as MACE, were also summarised as time-to-event variables and presented using the Kaplan-Meier method.
If a revascularisation procedure involving the treated lesion was performed before the planned 6-month repeat angiography, the last angiogram obtained before the re-intervention was used for the angiographic endpoint evaluation. The MACE per patient was ranked according to the highest category on a scale ranging from (1) cardiac death, (2) MI, (3) CABG to (4) TLR. Only events adjudicated by the clinical event committee were taken into account in the analysis of MACE.
All listed authors participated in the study design, enrolment of patients, and/or data interpretation. The data was processed and analysed by Cardialysis B.V., an independent clinical research organisation in Rotterdam, The Netherlands. The authors of this manuscript had full access to the data.
Results
Baseline and procedural characteristics
Between 3 July 2004 and 7 January 2005, one hundred and three patients were enrolled in the study. Baseline and procedural characteristics are shown in Table 1 and Table 2.
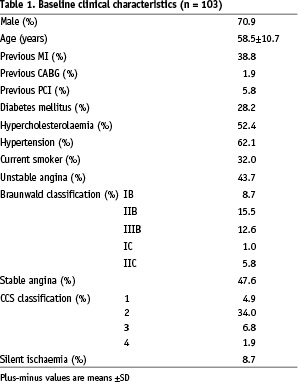
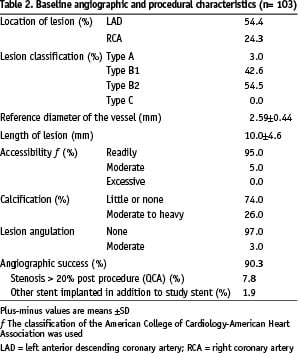
Overall, 71% of the patients were men, and the mean age was 58.5 years, with the expected prevalences of dyslipidaemia, diabetes, hypertension, and current tobacco use. Stenting was performed because of unstable angina in 43.7% of the patients (7% had post infarct angina). The target vessel was the left anterior descending coronary artery in 54.4% of the patients, the right coronary artery in 24.3%. Nearly all the treated lesions were class B1 or B2 according to the American College of Cardiology/American Heart Association classification. None were type C lesions. Although all the target index lesions were primary lesions, 1.9% of the patients had undergone previous coronary artery surgery and 5.8% had undergone previous percutaneous interventions for the treatment of other lesions.
The angiographic success rate was 90.3%. Eight percent (n=8) of the patients with unsuccessful angiographic success had a final residual stenosis > 20% (by off-line QCA) and 2% (n=2) needed more than one study stent. The procedural success rate was 87.4% as 3 patients experienced a MACE during hospital stay; all were related to peri-procedural myocardial necrosis (non-ST-segment elevation ACS). There were no cases of a clinically driven target lesion revascularisation procedure (CABG or PCI). No patient needed a platelet glycoprotein IIb/IIIa inhibitor during peri-procedural treatment at the index hospitalisation.
Quantitative Coronary Angiographic (QCA) analysis
Figure 2 shows the cumulative frequency of stenosis immediately after the index procedure and at six months.
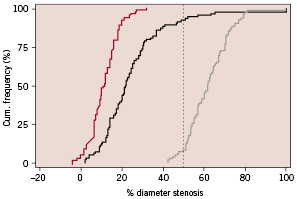
Figure 2. Cumulative frequency of % diameter stenosis.
Angiographic measurements are shown in Table 3.
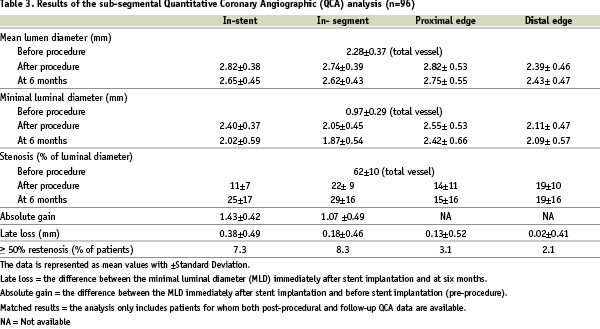
Angiographic follow-up at 6 months was obtained for 96 patients (93%), at a mean of 183±19.3 days (ranging from 119 to 296 days, including early symptomatic interventions). QCA revealed in-stent and in-segment late loss of 0.38±0.49 mm and 0.18 ± 0.46 mm, resulting in binary restenosis rates of 7.3% and 8.3%, respectively.
Major adverse cardiac events (MACE)
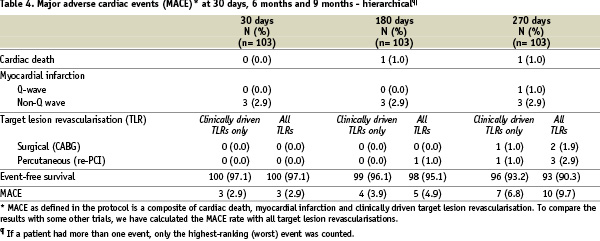
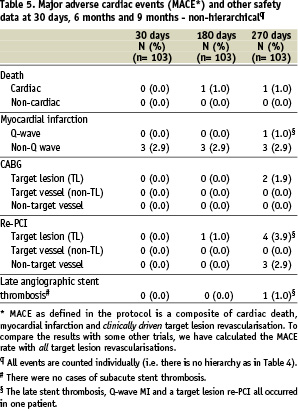
Ninety-eight percent of the patients completed nine months of clinical follow-up. One patient died and one patient refused further follow-up. Major adverse cardiac events (MACE) and other safety data are listed in Tables 4 (hierarchical count) and 5 (total count).
The Kaplan-Meier estimate of event-free survival is shown in Figure 3.
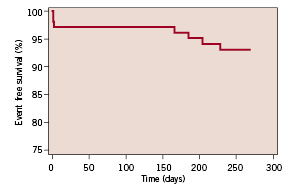
Figure 3. Kaplan-Meier estimate of the MACE-free survival rate.
The overall event-free rate (based on hierarchical MACE) at nine months was 93.2%, i.e. the overall rate of major cardiac events was 6.8%. There was one case of cardiac death: A 61 year old female patient suffered a cardiac arrest during transportation to the hospital with complaints of sudden breathlessness, general discomfort and diarrhoea. Three patients had a non-ST-segment elevation myocardial infarction at the time of stenting, resulting in a procedural success rate of 87.4% and a hierarchical MACE at 30 days of 2.9%. One patient suffered an ST-segment elevation myocardial infarction due to a late stent thrombosis 14 days after stopping clopidogrel and while on aspirin anti-platelet treatment only and underwent a successful primary repeat percutaneous coronary intervention (non-clinically driven re-PCI). In addition to this clinically-driven target lesion re-PCI, there was one clinically-driven target lesion coronary artery bypass grafting (CABG).
To compare the MACE results with those of some other trials, the MACE rate was also calculated with all target lesion revascularisations, clinically driven or not. Up to 270 days, three patients experienced a non-clinically driven TLR. This resulted in an overall event-free MACE rate of 90.3% at 9 months, i.e. a MACE rate of 9.7% (see also Tables 4 and 5).
Discussion
This multi-centre, prospective registry study reported on the efficacy and safety of the Infinnium™ stent for the treatment of single de novo lesions in a low to moderate risk CAD patient population. The concordant improvements in angiographic and clinical indices of restenosis at 6 months follow-up provide proof of principle that the current biodegradable-polymer-based Infinnium™ stent assembly reduces neointimal hyperplasia after coronary stent implantation, resulting in reduced rates of target lesion revascularisation. The QCA results at 6 months reveal a late loss that is in line with the observations in TAXUS I, II and IV trials.16,17 The MACE rate including all target lesion revascularisations of 9.7% at 9 months is acceptable and comparable with similar DES systems.16,17,29 The MACE included one case of cardiac-related death (at day 167) and one case of repeat PCI for an acute myocardial infarction due to late stent thrombosis (1%), 14 days after stopping clopidogrel and while the patient was on aspirin.
The achievement of a hospitable relationship between stent, coating matrix, drug, and vessel wall is extremely challenging. The long-term outcome of treatment with Infinnium™ paclitaxel-eluting coronary stent will reflect the response to all three components. The platform release kinetics are crucial. The hydrophobic properties of paclitaxel and its narrow toxic-therapeutic window require tightly controlled drug release. In the Infinnium™ paclitaxel-eluting stent, the drug is blended into a unique biodegradable polymeric matrix which is easily degraded into H2O and CO2 and readily eliminated from the body providing excellent biocompatibility. The concentration of drug on each stent was maintained at 1.4 µg/mm2 (versus 1.0 mm for the Taxus stent). Three different layers of combined drug and biodegradable polymers, each with a different release profile, were coated on the stent (coating thickness 4-5 µm), showing a bimodal release pattern with 50% drug release after 9 days, 90% after 38 days and 100% after 7 weeks.
The Infinnium™ is the first indigenously designed and evaluated “low cost” DES from Asia to have obtained a Conformité Européenne (CE) quality certificate for commercialisation in Europe. It holds strong promise for improving outcomes while limiting health care expenditure in CAD patients.
Limitations of the present study evaluating the Infinnium™ stent are the need for longer-term follow-up and the focus on standard-risk, focal, de novo lesions that may not reflect the more complex lesions and patients encountered in real-world practice. Whether the positive results in these patients can be expected across the full spectrum of patient complexity remains to be determined.
In conclusion, in patients with symptomatic ischaemic heart disease, treated for single, primary lesions in native coronary arteries, the low cost Infinnium™ stent system proved both effective in reducing restenosis at 6 months and safe with an acceptable rate of cardiac events at 9 months.
Appendix
Steering Committee
– D.S. Gambhir (PI), Department of Interventional Cardiology Kailash Hospital, Noida, India
– P.W. Serruys (Co-PI), Department of Interventional Cardiology, Thoraxcenter, Erasmus Medical Centre, Rotterdam, The Netherlands
– M. Doshi, Sahajanand Medical Technologies Pvt. Ltd.
– J. Symons, Cardialysis B.V., Clinical Research Organisation & Core Laboratories, Rotterdam, The Netherlands
Participating investigators/centres
– J.E. Sousa, Instituto Dante Pazzanese de Cardiologia, Sao Paulo, Brazil
– E. Ribeiro, INCOR Hemodinamica, Sao Paulo, Brazil
– D.S. Gambhir, Kailash Hospital and Research Center, Noida, India
– S. Dani, SAL Hospital & Medical Institute, Ahmedabad, India
– A. Dhar, Peerless Hospital & B.K. Roy Research Center, Kolkata, India
– J. Dalal, Lilavati Hospital & Research Center, Mumbai, India
– K.N. Vijava, Vijaya Heart Foundation and Research Center, Noida, India
– P.W. Serruys, Erasmus Medical Center, Rotterdam, The Netherlands
Funding/Support
This work was supported by a grant from Sahajanand Medical Technologies Pvt. Ltd.
Data Safety Monitoring Board
– A. Abhyankar, BDM Mahavir Cardiac Institute, Surat, India
– A. Vasavada, Sahrudya, Surat, Gujarat, India
– D. Desai, Hridayam, Surat, Gujarat, India
Clinical Event Committee
– T. Slagboom, Onze Lieve Vrouwe Gasthuis, Amsterdam, The Netherlands
– E. Ronner, Medisch Centrum Rijnmond-Zuid, Rotterdam, The Netherlands
– A.A. van den Bos, Amphia Ziekenhuis, Breda, The Netherlands
– C. Hanet, Cliniques Universitaires Saint-Luc, Brussels, Belgium
Data management and QCA Core Lab
Cardialysis B.V., Clinical Research Organisation & Core Laboratories, Rotterdam, The Netherlands.
Site monitoring
Cardialysis B.V. for Europe
Sahajanand Pvt. Ltd. for Brazil and India
