Funding sources: This study was sponsored by Guidant Corporation.
Stent, eluting stent, everolimus, randomized trial.
Abstract
Background: Everolimus is a sirolimus analogue with similar efficacy in animal models, and has been previously successfully tested in humans using an erodable polymer.
Methods: This first-in-man single blind multi-centre randomized controlled trial assessed the safety and efficacy of everolimus eluting from a durable polymer on a cobalt chromium stent in patients with de novo native coronary artery lesions. Sixty patients were allocated to stent implantation with an everolimus-eluting stent (n=28) or an identical bare stent (n=32). Patients had either stable, unstable angina or silent ischaemia. Suitable lesions treated were single de novo native coronary lesions with 50-99% stenosis and could be covered by a 18 mm stent. The primary endpoint was in-stent late loss at 180 days, analysed on a per treatment basis. The major secondary endpoint was percent in-stent volume obstruction (%VO) as measured by intravascular ultrasound (IVUS) at 180 days. The clinical secondary endpoint was major adverse cardiac events (MACE) at 180 days.
Results: At 6 months, (matched pairs angiographic analysis), the in-stent late loss, percentage diameter stenosis and percentage of patients with binary restenosis were 0.10 mm, 16% and 0% respectively, in the everolimus arm (n=23), as compared with 0.87 mm, 39% and 25.9%, respectively in the bare stent arm (n=27, p<0.001 for late loss and diameter stenosis, p = 0.01 for restenosis). Significantly less neointimal hyperplasia was observed in the everolimus group compared to the bare stent group (10 ± 13 mm3 vs 38 ± 19 mm3, p<0.001) and similarly, less volume obstruction (8.0 ± 10.4% versus 28.1 ± 14.0%, p<0.001). A major adverse cardiac event occurred in 2 patients in the everolimus arm versus 6 in the bare stent arm.
Conclusion: Everolimus eluted from a durable polymer on a cobalt chromium stent effectively suppresses neointimal growth at 6 months compared to an identical bare stent.
Introduction
Recent studies that have evaluated the local application of anti-proliferative drugs (sirolimus and paclitaxel) for the prevention of restenosis via a stent delivery system have shown that these therapies successfully inhibit the development of neointimal hyperplasia1,2.
Everolimus is an effective anti-proliferative agent3. On a molecular level, everolimus forms a complex with the cytoplasmic protein FKBP12. In the presence of everolimus, the growth factor-stimulated phosphorylation of p70 S6 kinase and 4E-BP1 is inhibited. The latter proteins are key proteins involved in the initiation of protein synthesis. Since phosphorylation of both p70 S6 kinase and 4E-BP1 is under the control of mammalian Target Of Rapamycin (mTOR), this finding suggests that, like sirolimus, the everolimus-FKBP12 complex binds to and thus interferes with its function. Disabling mTOR explains the cell cycle arrest at the late G1 stage caused by everolimus and sirolimus.
The feasibility of using everolimus on a drug eluting stent was determined by the FUTURE I trial4. This trial utilized an S-stent and bio-absorbable polymer system (both Biosensors International, Singapore) and confirmed the safety of the everolimus-eluting stent at 6 and 12 months. At 6 months, a 7.7% Major Adverse Cardiac Event (MACE) rate was observed with no thrombosis and no late incomplete apposition. The efficacy was demonstrated by significant reduction of in-stent tissue proliferation at 6 months: both angiographic in-stent late loss and IVUS% neointimal volume were reduced by 87%. No angiographic in-stent binary restenosis was observed in the everolimus-eluting stent arm. The 12 month FUTURE I results showed sustained safety and efficacy with no new MACE events, no aneurysms, no late stent malapposition, and no thrombosis observed between 6 and 12 months. Minimal Lumen Area and Luminal Volume Index were maintained up to 12 months and no in-stent binary restenosis was observed up to 12 months.
The SPIRIT First clinical trial represents the first clinical evaluation of the Guidant XIENCE™ V Everolimus Eluting Coronary Stent System (XIENCE™ V Everolimus Eluting CSS), to investigate the potential benefits of the local application of everolimus in a durable polymer in combination with a thin strut cobalt chromium stent.
Methods
Patient selection
This randomized single-blind trial was performed at 9 medical centers and enrolled patients from December 2003 to April 2004. It was approved by the ethics committee at each participating institution, and all patients gave written informed consent.
Patients were eligible for the study if they were aged above 18 years and had received a diagnosis of stable or unstable angina or silent ischaemia. Additional eligibility criteria were the presence of a single primary de novo coronary lesion that was 3.0 mm in diameter as assessed by on-line QCA, that could be covered by an 18 mm stent, a stenosis of between 50-99% of the luminal diameter, and a Thrombolysis In Myocardial Infarction (TIMI) flow grade of 1 or more. Patients were not eligible for enrollment if they had an evolving myocardial infarction, stenosis of an unprotected left main coronary artery, an ostial location, located within 2 mm of a bifurcation, a lesion with moderate to heavy calcification, an angiographically visible thrombus within the target lesion, a left ventricular ejection fraction of less than 30%, were awaiting a heart transplant, or had a known hypersensitivity or contraindication to aspirin, heparin, clopidogrel, cobalt, chromium, nickel, tungsten, everolimus, acrylic and fluoro polymers or contrast sensitivity that could not be adequately pre-medicated.
The Everolimus-eluting stent
The Guidant XIENCE™ V Everolimus Eluting CSS is comprised of the Guidant MULTI-LINK VISION® Stent and delivery system, and a drug eluting coating. The Guidant MULTI-LINK VISION® Stent is a balloon expandable stent, which consists of serpentine rings connected by links fabricated from a single piece of medical grade L-605 cobalt chromium alloy.
Everolimus is blended in a nonerodable polymer (this drug layer was coated over another nonerodable polymer primer layer). This coating includes of acrylic and fluoro polymers, both approved for use in blood contacting applications. This layer of everolimus-polymer matrix with a thickness of 5-6 microns is applied to the surface of the stent and is loaded with 100 micrograms of everolimus per square centimeter of stent surface area with no top coat polymer layer. The stent is designed to release approximately 70% of the drug within 30 days after implantation.
Everolimus (Certican®, Novartis Corporation) has been evaluated in clinical trials in the US and Europe for use as an immunosuppressant following cardiac and renal transplantation5. Everolimus has received market approval in the European Union.
Study procedure
Following the confirmation of angiographic inclusion and exclusion criteria and prior to the procedure, patients were allocated through a telephone randomization service and assigned in a 1:1 ratio to either an everolimus eluting stent or bare metal stent. A single stent 3.0 mm in diameter, 18 mm long was used in the study.
Lesions were treated using standard interventional techniques with mandatory pre-dilatation and stent implantation at a pressure not exceeding the rated burst pressure. Due to packaging differences, physicians were not blinded to the device. Post-dilatation was allowed with a balloon shorter than the implanted stent. In the event of a dissection occurring at the edge of the implanted stent, it was recommended that a single additional bare Guidant MULTI-LINK VISION® stent be implanted as animal data only on single everolimus stent implantation were available at the onset of the study; these patients were a priori excluded from the per-treatment analysis but are part of the acute success population. IVUS was performed after angiographically optimal stent placement had been obtained and was repeated if additional post-dilatation was performed.
Intravenous boluses of heparin were administered according to local standard practice. Treatment with aspirin, at a minimum dose of 80 mg per day, was started at least 24 hours before the procedure and continued indefinitely. A loading dose of 300 mg of clopidogrel was administered 24 hours before the procedure, followed by 75 mg daily for three months. Treatment with ticlopidine was permitted in case of clopidogrel hypersensitivity. Device success was defined as a final in-stent diameter stenosis of less than 50 percent by QCA using the assigned device. Clinical success was defined as the successful implantation of any device, with stenosis of less than 50 percent of the vessel diameter by QCA and no major cardiac events during the hospital stay.
Follow-up
Patients were evaluated at 30 days and 6 months. Further evaluations will be performed at 9 months and 1 year, with annual evaluations out to 5 years. At outpatient visits, patients were asked specific questions about the interim development of angina according to the Canadian Cardiovascular Society classification of stable angina. They were also monitored for MACE. Angiographic and IVUS evaluations were performed at 6 months, and will be repeated at 1 year. Prior to performing a follow-up angiogram, the physician was required to record in the source documents whether a revascularization (if required) was clinically indicated – defined as the presence of ischaemic symptoms and/or a positive functional ischaemia study.
Quantitative coronary angiography evaluation
Quantitative coronary angiography was performed using the CAAS II analysis system (Pie Medical BV, Maastricht, Netherlands). In each patient, the stented segment and the peri-stent segments (defined by a length of 5 mm proximal and distal to the stent edge) were analyzed. The following QCA parameters were computed: computer-defined Minimal Luminal Diameter (MLD), reference diameter obtained by an interpolated method, and percentage diameter stenosis. Binary restenosis was defined in every segment as diameter stenosis >50% at follow-up. Late loss was defined as the difference between MLD post-procedure and MLD at follow-up. Results are presented as matched pairs in the manuscript and as unmatched pairs in the Appendix. Unmatched pairs data is most commonly presented and utilises the mean QCA results of all projections obtained. Matched pairs data is more accurate as it compares the same views post-procedure and at follow-up and uses only QCA data of identical projections.
Intravascular ultrasound analysis
Post-procedure and follow-up stented vessel segments were examined with mechanical or phased array intravascular ultrasound using automated pullback at 0.5 mm per second. The coronary segment beginning 5 mm distal to and extending 5 mm proximal to the stented segment was examined. A computer-based contour detection program was used for automated 3-D reconstruction of the stented and adjacent segments. The lumen, stent boundaries and external elastic membrane (vessel boundaries) were detected using a minimum cost algorithm. The Stent Volume (SV) and Lumen Volume (LV) were calculated according to Simpson’s rule. The intra-stent neointimal volume was calculated as the difference between SV and LV. The percentage obstruction of the stent volume was calculated as intra-stent neointimal volume/stent volume*100. Feasibility, reproducibility and inter- and intra-observer variability of this system have been validated in vitro and in vivo6. Incomplete apposition was defined as one or more stent struts separated from the vessel wall with evidence of blood speckles behind the strut on ultrasound, while late incomplete apposition was defined as incomplete apposition of the stent at follow-up which was not present post-procedure.
Study endpoints
The primary angiographic endpoint was in-stent luminal late loss, as determined by quantitative angiography. Secondary endpoints (QCA and IVUS) at 6 months and 1 year included the in-stent and in-segment late loss, angiographic binary restenosis rate, percentage diameter stenosis; and in-stent percentage volume obstruction. In-stent was defined as within the margins of the stent while in-segment was defined as located either within the margins of the stent or 5 mm proximal or distal to the stent. Late loss was calculated as the difference between the follow-up and post-procedure minimum luminal diameter. Secondary clinical endpoints were a composite of major cardiac events, including cardiac death, Q-wave or non-Q-wave myocardial infarction, clinically driven surgical or percutaneous revascularization of the target lesion (MACE) or vessel (Target Vessel Failure) at 30 days, 6 months, 9 months, and annually up to 5 years after the index procedure; and acute device, procedure and clinical success. All deaths that could not be clearly attributed to another cause were considered cardiac deaths. A non-Q-wave myocardial infarction was defined by an increase in the creatine kinase level to more than twice the upper limit of the normal range, accompanied by an increased level of creatine kinase-MB, in the absence of new Q waves on electrocardiography.
The endpoints were adjudicated by an independent clinical events committee. In addition, a data and safety monitoring board that was not affiliated with the study sponsor reviewed the data to identify any safety issues related to the conduct of the study.
Statistical analysis
The primary endpoint and all trial endpoints were analyzed on the per-treatment evaluable population which consisted of patients who had no bailout stenting and no major protocol deviations, as evaluated in a blinded manner. Acute success was analyzed on the entire patient population.
The sample size for the study was determined based on the primary endpoint of in-stent late loss at 180 days and on the following assumptions: a single comparison of active to uncoated; one-tailed t-test, unequal and unknown variances in the two groups being compared; α=0.05; true mean difference between the bare stent group and the treatment group of 0.48 mm. This assumption was made based on the results of the VISION Registry (mean late loss=0.83 mm)7, SIRIUS trial (mean late loss=0.17 mm)8 and TAXUS IV trial (mean late loss=0.39 mm)9. (Assume the true mean late loss for the treatment group is 0.35 mm, the difference between the bare stent group and treatment group is calculated as: 0.83 mm - 0.35 mm = 0.48 mm). The standard deviation was assumed to be 0.56 mm in the bare stent group and 0.38 mm in the treatment group (based on the results of the VISION Registry study and SIRIUS trial); approximately 20% rate of lost to follow-up or dropout; approximately 10% of patients with bailout stents. Given the above assumptions, 30 patients per arm (with the analysis of 22 evaluable patients per arm) will provide 95% power for comparison. Although the trial was not powered based on the major secondary endpoint, percent volume obstruction at 180 days, enrolling 30 patients per arm (analysis of 22 patients per arm) would provide more than 96% power.
Binary variables were compared using Fisher’s Exact test. For continuous variables, means and standard deviations were calculated and groups compared using the Wilcoxon Rank-Sum test, except for the primary endpoint which was evaluated with a one sided t-test. Final 6-month results are presented in the manuscript, while the Appendix contains results that were available at the time that the 180-day report was prepared.
Results
Patient characteristics
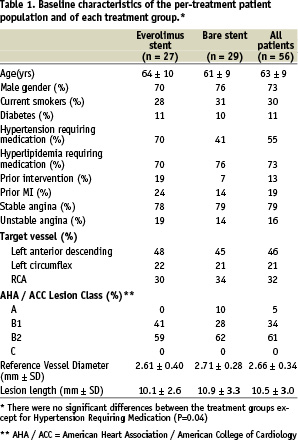
Between December 2003 and April 2004, 28 patients were randomly assigned to receive the everolimus-eluting stent, and 32 were assigned to receive the bare stent. As defined in the protocol, all results (except acute success) are presented for the per-treatment population (27 patients in the everolimus group, and 29 patients in the bare stent group, Figure 1). In the everolimus group there was one bailout procedure, and in the bare stent group there were two bailout procedures and one major protocol deviation (the patient was on the heart transplant waiting list). With the exception of a significantly higher number of patients with hypertension requiring treatment in the everolimus group, the two groups were similar with respect to clinical variables examined (Table 1).
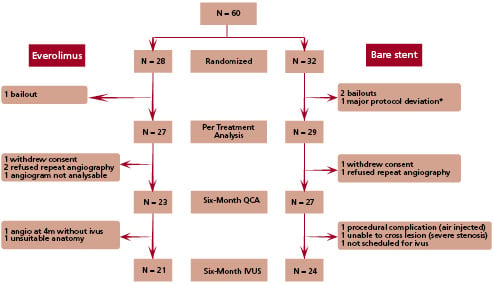
Fig. 1: Flowchart of patients
Procedural characteristics
The lesions in the two groups were treated similarly with the use of conventional techniques. Glycoprotein IIb/IIIa inhibitors, used at the investigators’ discretion, were administered to 7.4% of the patients in the everolimus group and 3.4% of those in the bare stent group. The two groups did not differ significantly with respect to the rate of device success (96.4% in the everolimus group and 93.8% in the bare stent group) or clinical success (96.4% in the everolimus group and 100% in the bare stent group).
Quantitative coronary angiography analysis
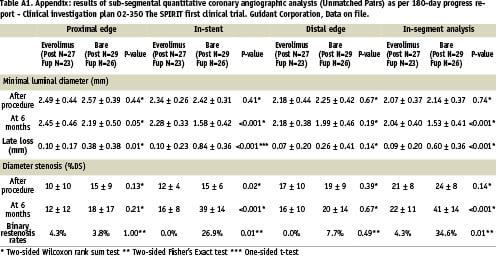
Angiographic data at 6 months were available for 50 of the 56 analysable patients (89.3%). The mean reference diameter of the target vessel, the mean length of the lesion at baseline, the reference vessel diameter and mean MLD of the stented segment were similar in the two groups (Tables 1 and 2). At six months, with matched pairs analysis, the mean MLD of the stented segment was significantly greater in the everolimus group. The mean in-stent late loss, percentage of stenosis, and percentage of patients with 50 percent or more stenosis were 0.10 mm, 16%, and 0%, respectively, in the everolimus group, as compared with 0.87 mm, 39%, and 25.9%, respectively, in the bare stent group (p<0.001 for late loss and diameter stenosis, p=0.01 for restenosis). Figure 2 shows the cumulative frequency of stenosis immediately after the index procedure and at six months in each treatment group. Table 2 and Figure 3 show the results of sub-segmental quantitative angiographic analyses for matched pairs. The late luminal loss at both the proximal and the distal edges of the stent was less in the everolimus group than in the bare stent group (p <0.01 for proximal and p=0.04 for distal). The late luminal loss in the stented segment was significantly less in the everolimus group than in the bare stent group (p <0.001).
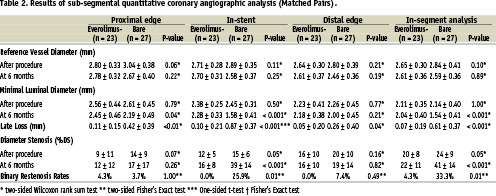
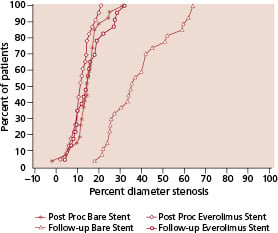
Fig. 2: Cumulative frequency of stenosis (in-stent) immediately after stenting and at six months
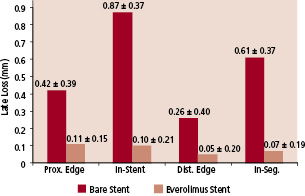
Fig. 3: Comparison of in-segment / in-stent late loss
Intravascular ultrasound evaluation
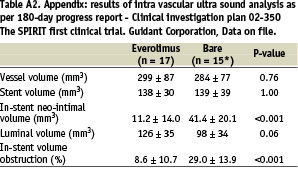
At six months follow-up, intravascular ultrasound evaluation showed no significant differences between the two groups with respect to the volume of the stent or the vessel volume (Table 3). Significantly less neointimal hyperplasia was observed in the everolimus-stent group compared to the bare-stent group (10 ± 13 vs. 38 ± 19 mm3, p<0.001) and similarly, significantly less volume obstruction, (8.0 ± 10.4% versus 28.1 ± 14.0%, p<0.001). Figure 4 is a cumulative curve of percentage volume obstruction. No in-stent volume obstruction was detected in almost half of the patients in the everolimus-stent group, whereas in the bare stent group, some degree of obstruction by neointima was present in all patients (Figure 4). No evidence of an “edge effect,” aneurysm formation, in-stent thrombosis, persistent dissection or late incomplete apposition were observed.
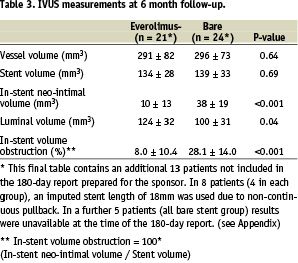
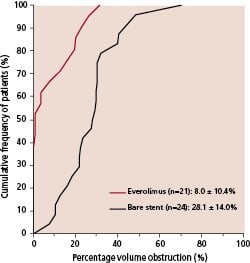
Fig. 4: Percentage in-stent volume obstruction versus cumulative frequency of patients. Values are expressed as mean ± standard deviation for each group.
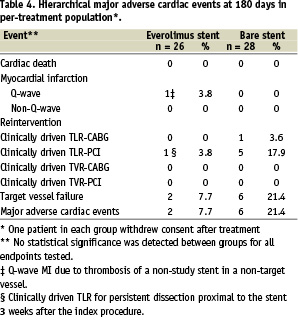
Major adverse cardiac events
Major adverse cardiac events are listed in Table 4. There was one Q-wave myocardial infarction in the everolimus group in a patient who underwent additional revascularization for angina in a non-target vessel 18 days after the study procedure and suffered thrombosis of this non-study stent 12 days later. The everolimus stent was patent with no evidence of thrombus at the time of the thrombotic occlusion of the non study stent. One patient in the everolimus arm underwent a clinically driven target lesion revascularization at 3 weeks for symptomatic persistent dissection at the proximal edge left untreated at the time of the procedure. There were no clinically driven target revascularizations in the everolimus group for restenosis. There were six clinically driven target lesion revascularizations in the bare stent group, five were treated percutaneously for restenosis and the sixth by bypass surgery. No adverse effects were attributable to everolimus or the polymer coating of the stents.
Discussion
The main finding of this randomized first-in-man study is that an everolimus-eluting stent coated with a durable polymer was associated with an in-stent angiographic late loss of 0.10 mm, significantly less than the corresponding bare cobalt chromium metal stent of 0.87 mm, which satisfied the primary endpoint of this trial and confirmed the efficacy of this system. Correspondingly, in-segment late loss was also significantly less in the everolimus-stent group.
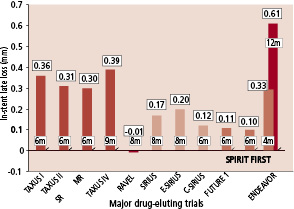
Currently, two different drug-eluting systems (sirolimus and paclitaxel) are available. Although no published scientific comparative data is to date available, it appears that, from historical randomized trials, a difference of approximately 0.2 mm in-stent late loss exists between sirolimus and paclitaxel. Even if the impact of restenosis and MACE is currently unknown, some slight difference in restenosis rates and MACE can be expected. New devices should at least equal the incumbents in performance. This performance may be judged on late loss, restenosis rate and / or the need for reintervention. With an in-stent late loss ranging from zero to 0.2 mm, it has been difficult to find a compound with the same efficacy, without resorting to the -limus family (Figure 5). With the sirolimus molecule being rather large and complex, it is therefore not surprising that major pharmaceutical companies have thoroughly explored its numerous analogues in order to develop a suitable competitor to sirolimus. The drug used in this study, everolimus differs from sirolimus by a substitution of a hydrogen radical/side-branch with a methyl sidechain.
Fig. 5: Comparison of in-stent late loss from drug-eluting trials.
The reason for developing new compounds is to improve on the side effects of the existing compounds such as delayed healing with re-endothelialization and fibrin11, early12 and late stent thrombosis13.
The success of the device lies in its three components - the drug, the polymer properties and the stent. The use of a sirolimus analogue is not in itself a guarantee of success since some of them have intrinsically, a potency in inhibition of up to 100 times less (e.g. tacrolimus), and some other analogues with equal in vitro inhibitory effects nevertheless fail to equally inhibit neointimal growth in vivo, because their duration of elution was suspected to be too short. However it has already been demonstrated that everolimus in clinical trials using a bioerodable polymer with a slower elution profile than sirolimus is effective in reducing late loss to below 0.2 mm4. Therefore the remaining challenge was to establish whether everolimus eluted from a durable polymer was also efficient and is addressed in this report.
Although the 6-month results are promising, one year angiographic and IVUS follow-up results are awaited to confirm the long-term results of this device in light of recent findings regarding an increasing late loss seen with other devices over time.
At the time of the publication of RAVEL, it was argued that the restenosis rate of the bare stent was excessively high at 26%. Similarly, in the present trial the restenosis rate in the bare stent arm was 25.9%. Nevertheless, it must be emphasized that in both cases these restenosis rates correspond to the value predicted and derived from multivariate analyses including as determinant parameters vessel size, MLD post, incidence of LAD disease and diabetics. Of interest, the late loss of the bare stent groups in RAVEL and this study were similar, corresponding to their restenosis rates. This is at variance with the VISION registry, and publications on stent strut thickness, but may be explained by the mismatch in stent size and reference diameter.
This study was powered for late loss and not for clinical events, and it was not surprising that the 3 fold reduction in events failed to be statistically significant. At the time of trial design, safety studies with overlapping eluting-stents in animal models had not been completed, requiring the use of bare stents for bailout. As a result of this confounder, these patients were a priori excluded from the per-treatment analysis. This study was however designed as a first in man trial with everolimus on an untested new durable polymer in combination with a cobalt chromium stent.
Acknowledgements
The authors would like to acknowledge the invaluable assistance of Susan Veldhof, RN, Cecile Dorange, MSc, Sophie Henry, MSEE from Guidant Europe, Diegem, Belgium for the conduct and reporting of this study.
Appendix
Sponsor: Guidant Corporation, Santa Clara, California, USA.
Principal Investigator: Patrick W. Serruys (The Netherlands).
Executive Committee: P.W. Serruys (Principal Investigator and Chairman, Rotterdam, The Netherlands); Gary Johnson (Vice President of Regulatory Affairs/Clinical Research, Guidant Corporation); Stan Fink (Director of Clinical Research USA, Guidant Corporation).
Data Safety Monitoring Board (DSMB) - J.G.P. Tijssen, Amsterdam, The Netherlands; F.W.A. Verheugt, Nijmegen, The Netherlands; W. Wijns, Aalst, Belgium.
Clinical Events Committee (CEC) - J. Vos, Amphia Ziekenhuis, Breda, The Netherlands; B.J.W.M. Rensing, Sint Antonius Ziekenhuis, Nieuwegein, the Netherlands; C. Hanet, Clinique Universitaire de Saint-Luc, Brussels, Belgium.
Data management - Angiographic and IVUS core laboratories: Cardialysis BV, Rotterdam, The Netherlands; Data Coordination Centre and Site Monitoring: Guidant Europe, Diegem, Belgium.
The following investigators and institutions participated in the SPIRIT First trial:
Clinical sites: J.J. Piek, Academisch Medisch Centrum, Amsterdam, The Netherlands (18 patients); F.J. Neumann, Herzzentrum, Bad Krozingen, Germany (14 patients); P.W. Serruys, Thoraxcentre, Erasmus Medical Centre, Rotterdam, The Netherlands (5 patients); M. Wiemer, HZ Herzzentrum, Bad Oeynhausen, Germany (5 patients); A. Zeiher, Uni. Klinikum Frankfurt, Frankfurt, Germany (4 patients); E. Grube, Heart Center Siegburg, Siegburg, Germany (4 patients); J. Haase, Red Cross Hospital, Frankfurt, Germany (4 patients); L. Thuesen, Skejby Sygehus, Aarhus, Denmark (4 patients); C. Hamm, Kerckhoff Klinik, Bad Nauheim, Germany (2 patients).
* Two-sided Wilcoxon rank sum test ** Two-sided Fisher’s Exact test *** One-sided t-test