This editorial refers to “First-in-Human Study of the Endeavor ABT-578-Eluting Phosphorylcholine-Encapsulated Stent System in de novo Native Coronary Artery Lesions: Endeavor I Trial.” by Ian T. Meredith et al., published in this issue of EuroIntervention.
Both the sirolimus-eluting stent (SES) (Cypher™; Johnson & Johnson - Cordis Corporation) and paclitaxel-eluting stent (PES) (Taxus™; Boston Scientific) were granted approval for marketing use by the U.S. and European regulatory authorities based on data derived from randomized clinical trials of over 3,000 patients and from registries with over 10,000 patients. These studies demonstrated significant reductions in restenosis and decreased the need for repeat revascularization with a similar safety profile when compared to bare metal stents (BMS)1,2.
In addition, both drug-eluting stents (DES) accumulate long-term data which exceeds five years with SES and three years with PES. The clinical experience gained from over 2,000,000 stents implanted in humans for a wide range of indications in clinical practice is overall satisfactory and set high standards for any new DES program.
The “Endeavor Stent System” is a new program that comprises a new cytostatic, antiproliferative and immunosuppressive agent in the same class of drugs as sirolimus, (ABT-578), a phosphorylcholine polymer-based coating, and an established cobalt-alloy stent with thin struts (Driver’ stent). With these features, the Endeavor system attempts to demonstrate equivalency and non-inferiority to the SES and PES systems while the latter two have started the second round of head to head trials.
The Endeavor 1 study was the “early bird” in a series of studies with the Endeavor stent system which was designed to examine whether this stent is truly a competitive candidate to the currently approved DES systems being used. The main objective of the study was to gather preliminary information on safety and efficiency of the Endeavor stent3. The study exceeded the classical phase 1 registry design by the number of patients and participating centers and carried a unique feature of two quantitative coronary angiography (QCA) and intravascular ultrasound (IVUS) follow-up time points at 4 and 12 months, which enables the evaluation of the performance of the stent in terms of efficacy over a longitudinal axis of time.
The study’s main findings demonstrated safety with low cumulative incidence of major adverse cardiac events: 1% at 30 days, 2% at 4 and 12 months, and low binary restenosis rates: 2.1% at 4 months and 5.4% at 12 months3. The investigators should be congratulated on obtaining a high rate of compliance in the angiographic and IVUS follow-up at 4 and 12 months, which provided an interesting observation related to the progression of the neointima formation - with a near doubling of the in-stent late loss from 0.33±0.36mm at 4 months to 0.61±0.44 at 12 months.
This finding raises several questions: 1) Was the 4-month angiographic follow-up an adequate time point?; 2) Will the progression of neointima formation continue beyond 12 months and if so when it would plateau?; 3) What is the importance of this relatively high in-stent late loss at 12 months for the Endeavor stent if it does not correlate with the increase in clinical events?; and 4) Will the clinical data continue to hold in a more difficult subset of patients and lesions?
Traditionally, binary restenosis for BMS and DES were measured at 6-9 months. An exception to this was with the first in-man study performed on 45 patients with the SES4. The angiographic and IVUS indices, which were associated with extremely low in-stent late loss and intimal volume at 4 months, remained nearly unchanged at 12 months and have led to the initiation of RAVEL, a randomized clinical trial which was designed to look at the traditional 6-month clinical, angiographic, and IVUS follow-ups. In contrast, the Endeavor I study confirms that 4 months is not a sufficient time point to rely on the performance of the Endeavor stent system in terms of angiographic and IVUS outcomes and perhaps this will apply to other new DES systems in the pipeline. The motivation to select an early time point of 4 months was derived from the eagerness of the investigators and sponsors to have a sneak preview on the data and perhaps to initiate earlier the pivotal study. Investigators should be weary about this aggressive strategy because counting on the 4-month results could be a moving target since healing is not complete at this early time point. It may create uncertainty and confusion and eventually be misleading in terms of the assumptions of the restenosis and late loss rates to miss an adequate sample size and power calculations. Without knowing what the restenosis rate at 6-8 months will be, the data derived from the 4-month follow-up in not reliable. Therefore, it is recommended that for future phase I clinical studies, sponsors should use the classic follow-up time frame of 6-8 months. This also may answer the question of why the in-stent late loss and binary restenosis rates nearly doubled from 4 to 12 months.
This progression of the in-stent late loss between 4 and 12 months reported in the Endeavor I study is a concern, and only further angiographic follow-up at a later time point can definitively address the question of when the progression of the neointima formation ceased. Data from Endeavor II, a pivotal study conducted in relatively more difficult subset of patients and lesions when compared to Endeavor I, suggests that the in-stent late loss at 8 months was 0.62 ±1.03 mm. Thus one can speculate that there is no more significant progression of neointima formation between 8 and 12 months and the majority of the healing process reached maturity at 8 months as previously shown with BMS and the PES. Yet, it would be warranted to address this question more scientifically in the Endeavor I population with additional angiographic follow-up time points at 18 or 24 months.
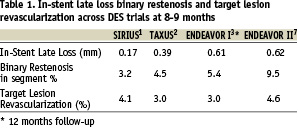
Continuous end points such as late loss have been used to test new stent technologies particularly in early-phase, small, sample size trials because of their inherent greater statistical power. Analysis of recent clinical trials demonstrated that the power to detect a treatment effect was greater for late loss than for binary angiographic restenosis5. As a result more and more trials are designed with a primary end point of in-stent or in-segment late loss rather clinical event. The Endeavor I results with low major adverse clinical events (MACE) at 1 year and the Endeavor II results7 with low target lesion revascularizations at 8 months, despite late losses of 0.61±0.44 and 0.62±1.03 mm respectively; challenge the utility of the late loss indices as a predictor to detect clinical events. Despite significant differences between the late loss indices in the Sirius1, Taxus2, and Endeavor I3 and II7 clinical trials, there were nearly identical event rates including target lesion and vessel revascularization across these trials. Perhaps with late loss indices of <0.6mm its ability to detect differences in clinical events is weak. (Table 1)
The Endeavor Stent System differs in stent material, design, polymer, and drug with respect to the approved SES and PES systems. The differences in potency is reflected by higher neointima volume obstruction and higher late loss measured by QCA and IVUS when compared to the SES system. Unlike the sustained results with the SES system from 4 to 12 months, the Endeavor system had progression of neointimal volume, which resulted in increased late loss. This can be a result of either drug difference, i.e. ABT 578 is an analogue of sirolimus, but is not as potent as sirolimus in terms of neointimal inhibition, or perhaps higher does are required to obtain a similar efficacy profile. The differences in efficacy can also be attributed to inferior kinetic release of the drug - the PC coating compared to the polymer with top coating of the Cypher system which enables more controlled release.
One of the advantages of the PC coating is its high biovascular compatibility. In animal studies, phosphorylcholine-coated stents have demonstrated significantly less platelet adhesion compared with uncoated stents6. If so, it carries the potential to be less thrombogenic when compared to other DES programs. In Endeavor I there was 1 event of stent thrombosis at <30 days. In both Endeavor I and II there was no evidence of late thrombosis >30 days, If it continues to show no late thrombosis in Endeavor III and IV, this stent will provide an important advantage over the currently approved DES programs and perhaps will not require prolonged regimens of antiplatelet therapy.
One of the disadvantages of the phase I clinical study is that it includes the “Vanilla” type of patients and lesions. For example, in the Endeavor I study only 16% of the patients were diabetic with a lesion length of 10.9±3.1mm and vessel diameter of 2.96±0.47mm. The real test for the Endeavor stent system will be when it is used in more complex patients and lesions. It is imperative that what is now considered borderline late loss will not continue to rise with the complexity of the patients and lesions.
Will the Endeavor I system be the new kid on the block? The results from the current study are encouraging especially with the announcement of the results of Endeavor II, which corroborate the results of Endeavor I in a more complex cohort of patients and lesions. By the time this editorial will be in press the results of Endeavor III will be available and Endeavor IV will be in the advanced stage of enrollment. The results of these two studies, conducted in a more complex cohort of patients in the U.S., and the long-term data from Endeavor I and II, and the continued absence of late thrombosis will determine whether the Endeavor stent system will be considered numero uno, first among equals, or equal among the existing SES and PES systems.
