Abstract
Aims: The Endeavor I study was the first clinical study evaluating the safety and feasibility of the Endeavor stent system in the treatment of symptomatic coronary artery disease. The Endeavor Stent System comprises a new cytostatic, antiproliferative and immunosuppressive agent in the same class of drugs as sirolimus, (ABT-578), a phosphorylcholine polymer-based coating, and an established cobalt-alloy stent with thin struts, (Driver™ stent).
Methods and results: One hundred consecutive patients with symptomatic ischemic heart disease due to de novo, obstructive lesions of native coronary arteries were treated with the Endeavor stent system at eight centers in Australia and New Zealand according to contemporary practice. The acute lesion, procedure, and device-deployment success rates were all 100%. Independent core laboratories analyzed quantitative coronary angiography (QCA) and intravascular ultrasound (IVUS) data immediately after stent implantation, and at 4 and 12 month follow-up. At 12 months, in-stent late lumen loss was 0.61±0.44 mm; in-segment late lumen loss was 0.43±0.44 mm, and neointimal hyperplasia volume was 14.2±11.8 mm3 (corresponding to a percent volume obstruction of 9.7%±8.5%). By QCA and IVUS, the pattern of neointimal hyperplasia was greatest within stent and not at the stent edges. The binary angiographic restenosis rate (defined as >50% diameter stenosis) at 4 and 12 months was 2.1% (2/97) and 5.4% (5/93) respectively. The cumulative incidence of major adverse cardiac events (MACE, defined as death, myocardial infarction, emergent cardiac surgery or repeat revascularization of the index lesion), was 1% at 30 days and 2% at 4 and 12 months.
Conclusion: This 100 patient pilot study demonstrates that the Endeavor stent system is a reliable and safe treatment for obstructive coronary disease, providing durable event free clinical outcomes to 12 months by suppression of neointimal proliferation of the target lesion. The results support further pivotal evaluation in larger randomized clinical trials.
Introduction
The use of endoluminal metallic stents became common practice during percutaneous coronary intervention after clinical trials indicated that stenting decreased restenosis rates when compared with conventional balloon angioplasty1,2. Restenosis rates in patients who receive coronary stents, however, can be as high as 20% to 40% after 6 months1-5. Stents coated with antiproliferative agents to inhibit neointimal hyperplasia represent a promising new technology for preventing restenosis. The use of sirolimus- or paclitaxel-eluting stents has been associated with significantly lower restenosis rates compared with conventional bare metal stents6-11. Mid and long term safety are still in question with late stent thrombosis attributed in part to polymer exposition and degradation or stent malapposition12-14. As the first generation of drug eluting stents has reduced the problem of restenosis with stenting, there is great interest in testing newer stent systems with greater flexibility that facilitates better deliverability to the target lesion.
A novel drug eluting stent platform, known as the Endeavor stent, combining the approved Driver™ chromium-cobalt-nickel alloy coronary stent system with an antiproliferative agent, ABT-578, and a biomimetic phoshorylcholine polymer, has been developed as a collaborative effort between Medtronic Vascular (Santa Rosa, CA., USA) and Abbott Laboratories (Abbott Park, Chicago, IL, USA). ABT-578 is a sirolimus analogue, that binds with a specific intracellular receptor FKBP12 to form a trimeric complex with the mammalian target of rapamycin (mTOR), thereby blocking mTOR’s signal transduction and inducing cell-cycle arrest in the G1 phase, limiting cell division15,16. The phosphorylcholine polymer, PC Technology, mimics a neutral and naturally occurring phospholipid. Clinical investigators have demonstrated the safety of PC coating in stent treated patients17-19. Also, stents coated with PC technology release drugs such as ABT-578 in a controlled and predictable fashion. The action of ABT-578, in combination with the favorable performance properties of the Driver™ stent, provide a novel and potentially valuable platform for the treatment of symptomatic patients with coronary artery disease.
The objectives of the ENDEAVOR I “first in human” multicenter prospective study of the Endeavor stent system were: (1) to assess the early feasibility and safety of implanting the Endeavor stent system in human coronary arteries, based on the device success rates and incidence of major adverse cardiac events (MACE) at 30 days; and (2) to assess the medium and long-term safety and efficacy of the Endeavor stent system, based on target lesion late lumen loss and neointimal hyperplastic volume, and the cumulative incidence of adverse events at 4 and 12 months.
Methods
The study was designed as a prospective, multi-center, non-randomized, single-arm study. One hundred subjects with symptomatic ischemic heart disease attributable to a native coronary artery stenosis, and amenable to treatment by percutaneous stenting, were enrolled in the ENDEAVOR 1 Trial from January 2003 through April 2003.
Data collection and core laboratories
All clinically relevant baseline and follow-up variables were collected on case report forms and processed by the Harvard Clinical Research Institute (HCRI, Harvard Medical School, Boston, MA). All data was independently monitored at the clinical site for validity. All clinical and coronary endpoints were determined by independent core laboratories (quantitative angiography, intravascular ultrasound, and ECG) and all clinical events were adjudicated by a coordinated independent clinical events committee at HCRI (non-involved cardiologists in the Boston medical area).
Quantitative coronary angiography (QCA). Coronary angiograms were obtained at baseline, at the completion of the stenting procedure, and at 4- and 12-month follow-up, and then submitted to the angiographic core laboratory (Brigham and Women’s Angiographic Core Laboratory, Boston, MA) for analysis. Quantitative angiographic measurements of the target lesion were obtained within the stent boundaries (in-stent measurements) and within the stent boundaries plus 5 mms proximal and distal to the stent (in-segment measurements). Reference and minimum lumen diameters and lesion length were determined using an automated edge-detection algorithm (Cardiovascular Measurement System, Medis; Medical Imaging Systems, Nuenen, The Netherlands20).
Intravascular ultrasound (IVUS). IVUS examinations were performed at the completion of the stenting procedure, and at 4- and 12-month follow-up. IVUS images were obtained from 40MHz Atlantis IVUS catheters (Boston Scientific, Natick, MA) during automated, motorized pullback at 0.5 mm/sec in accordance with guidelines provided by the IVUS Core Laboratory. Qualitative assessment and quantitative determinations of the areas and volumes of the vessels, stents, and lumens were made by the IVUS core laboratory (Cardiovascular Core Analysis Laboratory, Stanford University, Stanford, CA21).
Study patients
Study patients included single and multiple vessel coronary artery disease but only a single lesion per patient was treated in this trial. The target lesion must have been de novo, non-restenotic and in a native coronary artery. Lesions ≥50% and ≤100% diameter stenosis with a length ≤15 mm, in vessels with a reference diameter 3.0 mm to 3.5 mm, were included. Major exclusion criteria were a left ventricular ejection fraction <30%, significant (>50%) stenosis proximal or distal to the target lesion, occurrence of myocardial infarction (MI) within the preceding 72 hours, and a coronary interventional procedure of any kind within 30 days before or planned within 30 days after implantation of the stent system.
Clinical follow-up for all patients was performed at 30 days (telephone contact), 4 months (angiographic and IVUS measurements), 9 months (telephone contact), and at 12 months (angiographic and IVUS measurements). In addition, patients were consented to have yearly telephone follow-ups for 5 years from the date of the procedure. The study was conducted according to the Declaration of Helsinki, the medical ethical committees of all sites approved the protocol, and written informed consent was obtained from every patient.
Stent system
The Driver stent platform for the Endeavor stent system was CE mark approved in November 19, 2002 and received pre-market approval from the United States Food and Drug Administration in October 2003 for the treatment of coronary lesions. This cobalt-alloy stent has a low profile, and a strut thickness of 0.0036 inches (91 µm), designed to improve tracking and crossing in tortuous anatomy. In a prospective, multicenter, randomized registry study of 298 patients with symptomatic ischemic heart disease due to de novo or restenotic nonstented native lesions of a single vessel, the Driver™ stent platform was found safe and effective22.
The polymer, phosphorylcholine, is a synthetic copy of the predominant phospholipid in the outer membrane of red blood cells and has high biovascular compatibility. In animal studies, phosphorylcholine-coated stents have demonstrated significantly less platelet adhesion compared with uncoated stents19,23. In the SOPHOS (Study of PHosphorylcholine coating On Stents) trial, the phosphorycholine-coated BiodivYsio stent was found safe and effective for the treatment of native coronary artery lesions18.
The drug, a synthetic analog of sirolimus, ABT-578, binds with the intracellular protein FKBP-12 to form a trimeric complex with the mammalian target of rapamycin (mTOR), thereby blocking mTOR signal transduction, inducing cell-cycle arrest in the G1 phase, and limiting cell division15,16. The Endeavor stent system carries 10 µg of ABT-578 per millimeter of stent length. The drug is localized mainly on the ablumenal (arterial wall) side of the stent by means of the proprietary coating technique. For this study the stent system was available in a single length of 18 mm and in diameters of 3.0 mm and 3.5 mm.
Treatment procedure
Eight sites in Australia and New Zealand participated in this study. Stents were deployed according to a standardized procedure. Heparin was administered to maintain activated clotting time ≥250 sec or between 200 sec and 250 sec if a glycoprotein IIb/IIIa receptor inhibitor was administered. Patients received aspirin (300 mg to 325 mg daily), starting before the procedure, and clopidogrel (300 mg loading dose 6 hours before or at the procedure and 75 mg per day thereafter for 12 weeks). Pre-dilation was mandatory and undertaken with a balloon length shorter than the stent. Post-dilation, if required was also carried out with a balloon less than or equal to the stent length. Only 3.0 and 3.5 mm diameter Endeavor stents of 18 mm length were used in this study.
Endpoint definitions
Clinical endpoints were determined and summarized at each study follow-up milestone. Death was defined as the occurrence of any death during the study period. Myocardial Infarction (MI) was defined as any clinical event (peri-procedure or recurrent clinical presentation) in which there was evidence of myocardial necrosis (CK-MB >3 times normal), and further distinguished into Q- and Non-Q MI based on the development of Q waves in two or more contiguous lead on a standard 12-lead ECG. Target Lesion Revascularization (TLR) was defined as any percutaneous intervention or bypass surgery performed on the index target lesion at any time after the index procedure. Target Vessel Revascularization (TVR) was defined as any percutaneous intervention or bypass surgery performed on the index target vessel at any time after the index procedure, and Target Vessel failure (TVF) was defined as TVR or death or MI. Major Adverse Cardiac Events (MACE) was defined as TLR or death or MI. Stent thrombosis (ST) was defined as a thrombotic occlusion or stenosis of the stented target lesion, or in the absence of an angiogram, an ST-segment elevation MI in the territory of the index vessel.
Angiographic endpoints were determined at the baseline index procedure, and at 4 month and 12 month angiographic follow-up. Lesion length was measured as the length of contiguous coronary narrowing (defined as percent diameter stenosis >20%) containing both the target hemodynamically obstructive and adjacent non-obstructive plaque. Acute Gain was defined as the MLD immediately after the procedure minus the MLD at baseline. Late loss was defined as the MLD immediately after the procedure minus the MLD at 8-month follow-up. Percent diameter stenosis was defined as (1 - (MLD/reference vessel diameter) times 100). Binary angiographic restenosis (BAR) was defined as a >50% diameter stenosis at follow-up. Acute lesion success was defined as achievement of a target lesion percent diameter stenosis <50%); device success was defined as acute lesion success with the intended Endeavor stent; and procedure success was defined as device success without in-hospital death, myocardial infarction or emergent surgery.
Intravascular ultrasound (IVUS) endpoints were also assessed immediately after the baseline procedure, and at 4 and 12 months. Qualitative IVUS parameters used for safety evaluation included, stent edge dissection (defined as disruptions of plaque immediately adjacent to the stent ends where the flap could be clearly differentiated from the underlying plaque) and stent incomplete apposition (defined as ≥ 1 stent struts clearly separated from the vessel wall with evidence of blood speckle behind the strut).
Quantitative IVUS measurements were performed using commercially available planimetry software (Echoplaque, Indec Systems) according to previously validated and published protocols24. External elastic lamina (EEL), stent, and lumen area were manually traced for serial cross sections with the axial distance of nearly 0.5 mm throughout the stented segment. NIH area was computed as stent minus lumen area. Volumetric analysis was performed by means of Simpson’s method for the entire stented segment.
In detailed analysis, NIH growth was analyzed as a NIH area/volume divided by the corresponding stent area/volume to adjust for the different stent sizes. The entire in-stent NIH growth was evaluated as percent NIH volume (NIH volume x 100 / stent volume: % NIV). The most severe luminal obstruction by NIH growth within the stent segment was determined as the maximum of percent NIH area (NIH area x 100 / stent area: % NIA).
Primary Safety Endpoint. The primary safety endpoint was MACE at 30 days, analyzed on an intent-to-treat basis. Consented subjects who met eligibility criteria were analyzed regardless of the subsequent sequence of events.
Primary Efficacy Endpoint. The primary efficacy endpoint was angiographic in-stent late loss at 4 months, analyzed on an intent-to-treat basis. All eligible subjects who received a stent and follow-up angiography were included in the primary efficacy endpoint analysis.
Study objective and statistical analysis
The main objective of this study was to demonstrate the ease-of-use (feasibility) and acute safety of the ABT-578 Coated Driver Coronary Stent System while gathering preliminary information on mid- and long-term safety and efficacy of the device. The trial was therefore designed to minimize the number of subjects exposed to the device while still providing enough information for a preliminary indication of safety and feasibility. A sample size of 100 subjects was deemed sufficient to provide reasonable confidence in the estimates of safety and efficacy generated by this study.
Based on observations from contemporary stenting trials, the expected incidence of MACE at 30 days was estimated to be approximately 4%, reflecting a true event rate of 4 or fewer subjects in this pilot study. However, the random error of estimating a true 4% rate with 95 subjects (assuming 5% loss to follow-up at 30 days) is relatively large, and a statistical test designed to test the probability of a true rate greater than 4% allowed for sufficient alpha (type I) error. The upper 95% confidence bound for a 4% rate with n=95 was 7.9%. Thus, the proposed test for demonstration of acute safety and feasibility was observation of a 30-day MACE rate ≤ 7.9%, or observation of MACE in ≤ 8 subjects.
An in-stent late loss of approximately 0.8 - 1.0 mm±0.55 mm (mean±SD) has observed in contemporary bare metal stenting trials25. Assuming a 20% loss to follow-up, this trial had 89% power to detect a late loss difference of 0.2 mm or greater from 0.8 mm. These estimates were derived using a one group t-test with a two-tailed alpha value of 0.05.
Analysis and reporting of results. All analyses are made under intention-to-treat, defined as the set of all subjects with qualifying stenosis in which treatment with the Endeavor system was intended, i.e., all patients in whom a guidewire was used to initiate the procedure. Analyses of angiographic and IVUS data, collected at baseline, 4, and 12 months follow-up were analyzed on all patients with technically successful paired data for the pairwise comparisons. Categorical variables were tested using contingency tables analyses (exact or chi-square approximations), and continuous variables were tested using unpaired Student’s t-test or Wilcoxon rank-sum test, depending on variable distribution. All statistical tests were 2-sided, and a p-value <0.05 was considered statistically significant.
Results
Demographics. Baseline clinical characteristics for the 100 enrolled patients are shown in (Table 1) demonstrated a mean age of 59 years (range 35-76). The population was 79% men, and 47% of the subjects had a prior MI, while 16% had a history of diabetes mellitus. The age, sex distribution and risk factor profile were consistent with other contemporary study samples of patients presenting with ischemic symptoms due to a discrete, de novo lesion in a single, native coronary artery26.
Subjects and follow-up. Procedural and lesion characteristics (Table 2) were summarized for a total of 107 implanted stents in 100 patients; in seven patients two stents were implanted. Complete clinical follow up at 12 months was available for all patients. At 4 months, three patients were lost to QCA follow-up (97% follow-up) and 95 patients had technically successful IVUS (95% follow-up). At 12 months, nine patients were lost to QCA follow-up (91% follow-up) and 86 patients had technically successful IVUS (86% follow-up). Angiographic estimates at 12 months were made on 93 subjects: 91 who had actual 12 month follow-up plus two subjects whose 4 month data was carried forward because they underwent target lesion revascularization at 4 months.
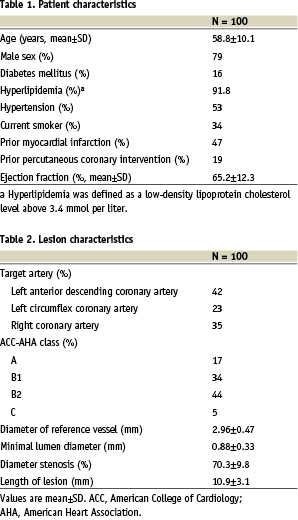
Clinical results. The acute lesion success, device success, and procedure success were all 100%. Table 3 summarizes the composite and component adverse event rates at 30 days, 4 months and 12 months. The in-hospital incidence of major adverse cardiac events (MACE) was 0%, while MACE at 30 days was experienced by one subject (1%, non-Q wave MI post procedure, day 10 associated with stent thrombosis and repeat revascularization via percutaneous intervention then bypass surgery). At 360 days, the cumulative MACE rate was 2% (one additional subject had a target lesion revascularization 112 days post procedure). Both the 30-day 1% MACE rate and the 12-month 2% MACE rate were well less than the tolerable upper safety boundary of 8% ≤ 8 patients) that was pre-specified in the clinical protocol. There were no deaths over the 12 month study period.
The Kaplan-Meier estimate of freedom from MACE at 12 months was 98% [95% CI: 94.1,100.0]. The cumulative incidence of TVF at 12 months was 2.0% (2/100). The TLR-free survival rate at 12 months post-procedure was also 98.0% and the cumulative incidence of TLR at 12 months was 2.0% (2/100).
Angiographic and IVUS results at 4 and 12 months follow-up. Figure 1,2 and Table 4 present the QCA results after the index stenting procedure at 4 months and 12 months after the index stenting procedure at 4 months and 12 months. Binary angiographic restenosis was seen in 2 of 97 patients (2.1%) at 4 months. In-segment late lumen loss at 4 months follow-up was 0.21±0.40 mm, and in-stent late loss was 0.33±0.36mm. The proximal and distal edge late lumen losses were 0.12±0.42mm and 0.09±0.36mm respectively at 4 months and 0.29±0.46mm and 0.21±0.36 mm at 12 months. At 12 months follow-up, five subjects had binary angiographic restenosis (5.4%), and in-segment late lumen loss was 0.43±0.44 mm, and in-stent late loss was 0.61±0.44 mm. The in-stent late loss of 0.61 mm was statistically lower than the 0.8 mm standard that was pre-specified (p<0.001), and also lower than the recently reported Driver platform which had an in-stent late loss of 0.93 mm22.
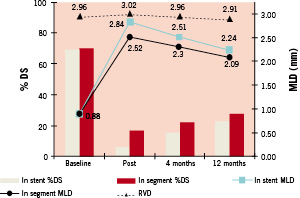
Figure 1. Quantitative coronary angiographic analysis of stent edge effect.
In-stent and In-segment minimum lumen diameter and% diameter stenosis at the 3 specified time points. Line plots reflect minimum lumen diameter (MLD), circles indicate in-segment MLD while squares refer to in-stent MLD. Triangles refer to the reference vessel diameter. Bar graphs indicate% diameter stenosis in-stent and in-segment respectively at each time point.
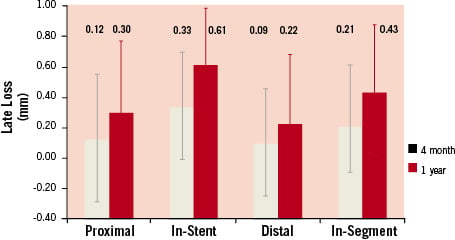
Figure 2. Bar graph showing the mean late lumen loss associated with implantation of the Endeavor stent at 4 and 12 months in the 5 mm segment proximal to the stent, in-stent, 5 mm segment distal to the stent an in-segment.
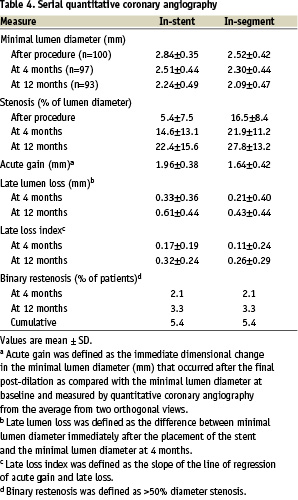
Table 5 presents the IVUS results immediately after the index stenting, at 4 months and at 12 months. Of the 99 IVUS examinations that were suitable for evaluation at the index procedure, 95 studies were evaluated at 4 months. IVUS examinations received in 2 patients were not analyzable and two further patients did not undergo IVUS, one as a result of reaching an endpoint. At 12 months, 86 IVUS studies were available for evaluation.
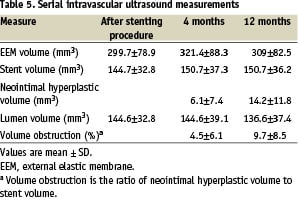
At the baseline IVUS procedure, 11 stent edge dissections were identified. Five were intimal edge dissections, 3 were medial edge dissections and a further 3 were treated with additional stents. At 4 months no edge dissections were identified and all 8 non-stented edge dissections had healed. Incomplete stent strut apposition was identified in 12 patients at the baseline study. Eight of these had resolved at 4 months and 4 were preserved incomplete appositions. Late acquired apposition was not seen in any patient at either 4 or 12 months.
IVUS derived minimal lumen cross sectional area was 6.2±1.4mm2 post procedure and 6.0±1.7 mm2 and 5.7±1.8 mm2 at 4 months and 12 months respectively. The neointimal hyperplastic volume was 6.1±7.4 mm3 and 14.2±11.8 mm3 at 4 and 12 months respectively. The percent neointimal volume obstruction was 4.5± 6.1% at 4 months and 9.7±8.5% at 12 months.
Discussion
In this study we report the first-in-human safety and efficacy of the Endeavor drug-eluting stent system comprising the Driver™ stent, phosphorylcholine PC technology coating and ABT-578, a sirolimus like analogue. To the best of our knowledge this is the first report of a DES system based on a new generation thin strut, Chromium-cobalt alloy stent.
The trial was comparatively large for a first in human study as the main objective was to demonstrate acute safety of the Endeavor stent system while gathering invaluable but preliminary information on mid-and long-term safety and efficacy. A sample size of 100 patients was calculated to be sufficient to provide reasonable confidence in the estimates of safety and efficacy and thus provide a strong platform on which to establish larger randomized clinical studies, namely the three planned (Endeavor 2, 3 and 4) multicentre pivotal randomized trials. In addition to its large size for a pilot study, it is the first to evaluate restenosis during the biological restenosis period, at 4 and 12 months. Moreover there were other technical specifications that insured the percutaneous procedure was aligned with best contemporary clinical practices, such as the requirement for pre and post dilation to be undertaken with balloons less than or equal to the stent length in accordance with data of the effect of balloon length on restenosis with drug-eluting stents25,27.
This study was conducted in 8 high volume centers performing more than 1000 procedures per year by operators performing at least 200 cases per year. Device, lesion and procedural success was achieved in 100% of patients. In part, the high success rates are likely a function of the excellent performance characteristics of the Driver stent and delivery system and the type of lesions and vessels treated.
Comprehensive clinical follow-up was achieved in all patients at 4 and 12 months underpinning the calculated power of the trial to provide a meaningful estimate of the true MACE rate. The proposed test for demonstration of acute safety and feasibility was observation of a 30 day MACE rate of <7.9% or observation of MACE in fewer than 8 patients. To this end, the primary safety endpoint of MACE at 30 days was successfully achieved with a 1% MACE rate. Mid and longer term clinical safety was sustained with a 4 and 12 month MACE rate of only 2%, that is one further MACE event between 30 days and 12 months. Angiographic follow up was achieved in 97% of patients at 4 months and 93% at 12 months. Repeat intravascular ultrasound assessment was achieved in 95 and 86% of patients at 4 and 12 months respectively. Excellent levels of patient retention in the invasive components of the follow up study insured this power was maintained.
We observed an instent late loss of 0.33±0.36 mm at 4 months with further progression to 0.61±0.44 at 12 months, and in segment late loss of 0.21±0.40 progressing to 0.43±0.44 at 12 months. This modest angiographic late loss was associated with a low cumulative binary restenosis of 5.4% at 12 months and low TLR (2%, which includes one patient with a stent thrombosis at 10 days, and thus only one patients had TLR for clinical restenosis) TVR and TVF (2%) rates.
Although not readily comparable due to differences in patient, vessel, lesion and procedural factors and the timing of follow up studies in-stent and in-segment late loss appear to be marginally greater than observed with Sirolimus8,28,29, Everolimus30 and Paclitaxel when delivered on a polymer coating6,31 in early phase clinical trials. However clinical endpoints such as MACE, TLR and TVF are at the very least similar if not better.
The present study also provides important insights into the nature, distribution and time dependence of neointimal proliferation within this stent system. We observed a modest decrement in minimal in stent lumen area (MLA) of approximately 0.5 mm2 over 12 months. This was similar in magnitude to the loss of MLA observed in the cohort of patients undergoing IVUS in the SIRIUS study between the baseline and 8 month interrogations32.
There was an approximate doubling of the neointimal hyperplastic volume and thus percent volume obstruction between 4 and 12 months in our study highlighting the importance of later angiographic and IVUS follow up in DES studies and the need to consider this when comparing studies with different and singular follow-up time points. At twelve months we observed a mean of 9.7% neointimal volume obstruction within the Endeavor stent system which is comparable to that observed in the IVUS follow up cohorts of TAXUS II6,33 and TAXUS IV but marginally greater than observed in Sirius, Future I and II and Ravel8,12,29,30,34. Importantly our IVUS studies confirmed appropriate healing of peristent dissections and no evidence of late acquired malapposition. There were no late (>30-day) subacute thromboses.
The present data provide a strong foundation for larger scale clinical trials of the Endeavor platform including the 1200 patient Endeavor II powered primarily for clinical endpoints. Such studies will provide greater insight into the benefit of this system in patients with somewhat more complex lesions.
Conclusion
The Endeavor stent system is safe and feasible with complete acute lesion, procedure, and device-deployment success. Treatment of patients with symptomatic ischemic heart disease and single-vessel de novo lesions, with the Endeavor stent system resulted in sustained clinical effectiveness and only modest changes in vessel lumen dimensions up to 12 months. The healing process translated in mild neointimal proliferation or late lumen loss but this needs to be prospectively tested to determine the optimum balance between biologic response and adequate clinical anti-restenotic properties.
The present Endeavor I, first-in-human study will be augmented by a comprehensive clinical research program of trials powered to meet clinical endpoints. Endeavor II is a multicenter, double blind, randomized study to evaluate the Endeavor stent system compared with the Driver™ bare metal stent in the treatment of 1200 patients with symptomatic ischemic heart disease and single-vessel de novo lesions, including lesions up to 27 mm in length and small vessels (≥2.25 mm reference vessel diameter). Endeavor III is a multicentre, double blind, randomized study to compare the Endeavor stent system with a sirolimus-eluting stent in the treatment of 436 patients. Randomization in Endeavor III will occur on a 2 to 1 basis; the primary endpoint is late lumen loss at 8 months. Endeavor IV is a multicentre, double blind, randomized study to compare the Endeavor stent system with a paclitaxel-eluting stent in the treatment of >1500 patients. The primary endpoint of Endeavor IV is target vessel failure at 9 months. The present study provides a sound and safe platform for these larger scale studies. We await their outcome with considerable interest.
Acknowledgements
The Endeavor I investigators also include Mark Adams, MBBS, Ph.D, Royal Prince Alfred Hospital, Sydney, Australia; Con Aroney, MBBS, The Prince Charles Hospital, Brisbane, Australia; Mark Pitney, MBBS, Eastern Heart Clinic, Randwick, Australia; Brigham and Women’s Hospital, Boston, MA; and Peter Zimetbaum, MD, of Harvard University, Boston, MA.
