Abstract
Aim: To study the feasibility, safety and efficacy of the Recover® LP 2.5 assist device in patients scheduled for high risk off-pump coronary bypass surgery, percutaneous coronary intervention or patients in cardiogenic shock.
Methods and results: 40 patients presenting with cardiogenic shock (n=13) or scheduled for a high risk revascularisation (n=27) were included.
36 were selected for safety and feasibility analysis. In 3 patients the pump could not be placed in an adequate position. 5 patients had access related complications. In 9 patients free Hb rose above 80 mg/dl. 3 malfunctions and early device-removal occurred. After device modifications these problems did not recur. CO in the shock group increased significantly: 4.4 l/min±1.9 to 4.8 l/min±1.2 (p=0.0178).
The left ventricular filling pressures decreased in both groups (22 mmHg±7.5 to 16 mmHg±6 in the shock group, [p=0.0008] and over 6 hours from 14.3 mmHg±5.8 to 10 mmHg±2.9 in the high-risk revascularisation group,[p=0.0327]).
Conclusions: The Recover® LP 2.5 micro axial pump allows, via percutaneous approach, partial unloading of the left ventricle. The technique is, after design modifications, feasible and safe and results in haemodynamic improvement.
Introduction
Since the introduction of off-pump coronary artery bypass surgery (OPCAB) and percutaneous coronary interventions (PCI) in patients considered to be at high-risk for haemodynamic collapse or high likelihood of haemodynamic collapse, the need for an easy to place left ventricle assist device has emerged. The device most commonly used in this setting is the intra-aortic balloon pump (IABP). Haemodynamic support with unloading of the left ventricle results in reduced filling pressures, decreased wall stress and oxygen demand. This lowers the risk for ischaemia and prevents subsequent haemodynamic instability1-14.
However, the IABP provides, by afterload reduction, only a limited increase in cardiac output and becomes less effective in patients with tachyarrhythmias and full-blown left ventricular dysfunction. Meanwhile substantial experience has been gained with more forceful assist devices14-18. In patients with cardiogenic shock, haemodynamic support results in improved organ perfusion, and when combined with interventions for reperfusion or revascularisation, it can result in increased survival rates19-27.
The Hemopump® (Medtronic®, Minneapolis, USA) and the Recover® (Impella®, Aachen, Germany) were the first axial pumps clinically used peri-operatively and for patients in cardiogenic shock28-30.
Based on the experience with Recover® 5.0, a smaller percutaneous transvalvular assist device, Recover® LP 2.5 (Figure 1) was developed.

Figure 1. Recover® LP 2.5, initial study (left) and market design (right).
It is a miniaturised pump, providing load depending flow unloading the left ventricle. The pump incorporates an impeller with 2 vanes driven by an electrical motor and has a 4 mm inflow cannula (12F). The pump is placed through the aortic valve and aspirates blood from the left ventricle cavity and expels it into the ascending aorta. The pump can be introduced via a femoral percutaneous approach using a 13 F sheath. The driving console allows 9 gradations in speed. At maximum speed (51.000 rotations per minute) a flow of 2.5 l/min is provided.
In this first trial in humans we studied the safety and efficacy of the Recover® LP 2.5 pump in patients scheduled for high-risk revascularisation (PCI or OPCAB) as well as in patients presenting with cardiogenic shock.
Methods
Patient population
The study was a multicentre (3 centres) non-randomised prospective trial to assess the safety, feasibility and the haemodynamic effects of the Recover® LP 2.5 in patients presenting with cardiogenic shock (group I) and in patients scheduled for a high-risk revascularisation (group II).
Haemodynamic criteria for cardiogenic shock were a mean arterial pressure (MAP) < 70 mmHg, a cardiac index (CI) <2.0 l/min and a pulmonary capillary wedge pressure (PCWP) >18 mmHg or patients had to be on inotropes to maintain the MAP >70 mmHg, a CI >2.0 l/min with a PCWP > 18 mmHg30. Patients could be on balloon pump support before Impella implantation.
The revascularisation procedure was considered to be high-risk, if at least left ventricular function was impaired. Based on the patients characteristics, the EuroSCORE was calculated31.
Device-related exclusion criteria were: anticipated femoral access problems, the presence of a known mural intracardiac thrombus, a ventricular septal defect, hypertrophic obstructive cardiomyopathy, aortic valve disease (stenosis and/or regurgitation or an artificial valve).
Exclusion criteria were: a body mass index (BMI) higher than 37 kg/m2, participation in another clinical investigation during the last 60 days, age less than 18 years of age, pregnancy, refusal of blood transfusion, shock due to volume depletion.
The Ethical Committees approved the study protocol. Patients gave written informed consent if their mental status permitted. Otherwise consent of a relative was obtained.
Implantation of the device
Femoral percutaneous access was obtained by Seldinger technique. In one centre prior to the introduction of a 13 F sheath, a Prostar XL closure device (Abbott®, Redwood City, USA) was placed, in order to obtain quick haemostasis at the time of pump removal. During the trial, a dedicated 13 F sheath was developed (Figure 2), it is a peal away sheath, with a separable valve.
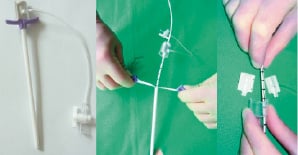
Figure 2. The peal away sheath with valve and clasp.
This valve is kept together by a clasp. After introduction of the 13 F sheath, a second 7 F sheath is introduced in the larger sheath, a Judkins Right (JR4) catheter is advanced over a J-tip wire. The aortic valve is crossed and a 0.014” wire advanced through the JR4 in the left ventricle. The use of a pigtail is avoided since the 0.014” wire can get trapped in the side holes. The 7 F sheath and JR4 catheter are removed, leaving the 0.014” wire in the left ventricle. After purging the pump with a heparinised glucose 30% (20-40%) solution and a test run, the pump is advanced through the aorta, over the 0.014” wire into the left ventricle.
This enables a forceless crossing of the aortic valve, preventing kinking of the cannula or catheter during placement which could lead to pump malfunction.
After wire removal, pump rotation is started. Speed is gradually increased under fluoroscopic guided positioning, in order to obtain a stable position without suction on the left ventricular wall or pump displacement in the aorta. During the trial, pump design modifications were performed to obtain a more stable position in the left ventricle cavity and to reduce the shear forces. The main adaptations were: 1. stiffening of the cannula, 2. adding a pigtail at the tip of the cannula, 3. increasing the gap between the impeller vanes and the pump housing. At maximal speed (51,000 rotations per minute) the pump provides 2.5 l/min blood flow.
For those patients in need of longer support times, the peal away sheath is removed and a tapered “access-closure” sheath (10-13 F), present on the shaft (Figure 3), advanced in the artery, until the bleeding stops.

Figure 3. The tapered sheath on the shaft.
This tapered sheath, together with arterial recoil can result in a smaller device remaining at the access site, reducing the risk of limb ischaemia.
Repositioning of the cannula can be performed without compromising sterility at the access site.
All patients received heparin via the lubrification system of the pump (glucose 30%). All patients received 40 IU/kg of heparin to achieve an ACT>250 seconds prior to pump implantation. Heparin administration was dosed to achieve an activated PTT between 50 and 80 seconds.
Biochemical markers
Blood count, renal function, liver function, lactate and free haemoglobin were measured at baseline, 1 hour after pump start and at 6, 12, 18, 24, 36 hours, 2, 3, 4, 5 days after arrival in the intensive care unit.
Haemodynamics
Monitoring of pump data i.e. performance level, pressure signal (mmHg), motor current (amperes), purge flow rate (ml/h) and purge pressure (mmHg), were registered every 10 minutes during implantation and later, once an hour. Continuous cardiac output (Vigilance Baxter®, Deerfield, USA), mean arterial pressure, mean pulmonary arterial pressure, mean capillary wedge pressure were measured each hour, and up to 6 hours after pump removal.
Endpoints
Predefined endpoints for safety were the absence of device-related limb ischaemia, neurological deficits and a free haemoglobin level exceeding 80 mg/dl.
Predefined endpoints for feasibility were ease of insertion, positioning and explantation (graded).
Efficacy end-points were a significant decrease in filling pressures, increase in cardiac output and increase in mean blood pressure.
Follow-up for clinical events extended to one year after pump explantation.
Sample size and statistical analysis
To achieve first experience and information about safety and feasibility in application of the Recover® LP 2.5 device, the total number of patients treated was limited to 40. The safety population consists of those patients fulfilling the major inclusion criteria, i.e. patients where it was initiated to apply the Recover® LP 2.5 device. This population was considered with respect to feasibility and safety questions. The efficacy questions (haemodynamic output) were only investigated in these patients where the pump did run.
Categorical variables are described by numbers and percentages. Further, 95% confidence intervals of the estimated percentages (Clopper and Pearson, 1934) were given. The measurements of the haemodynamic parameters (cardiac output and index, mean arterial pressure and pulmonary capillary wedge pressure) were not time-synchronized. To evaluate the time effect of measurements, a repeated measurement analysis of variance model was fitted to the data (SAS® software). The time effect was modelled as a linear regression variable. To describe the mean effect at certain time points, we used averaged measurements of the repeated observation within the time intervals and computed means and standard deviations (SD) represented as the value ± SD or standard errors (SE) of the resulting averages.
Results
From 02/2003 to 10/2004, 40 patients were screened in three centres, Leuven (n=23), Siegburg (n=7) and Maastricht (n=10). Cardiogenic shock patients were only included in Leuven (n=8) and Siegburg (n=3). Of these 40 patients, 1 patient withdrew consent and in 3 patients the surgeon decided not to place the device and to perform off-pump revascularization without mechanical support after the patient had been included in the study. 36 patients were included for the safety and feasibility analysis. In 3 of these 36 patients the pump was never activated, in 2 patients the pump did not cross the aortic valve, and in 1 patient the pump was displaced into the aorta and recross was impossible. Of the 33 remaining patients (included in the efficacy analysis) 11 were in cardiogenic shock (group I) and 22 underwent a high-risk revascularisation procedure (group II). Of the shock patients, 9 had an IABP before Impella implantation.
Demographics of the studied population are presented in table 1.
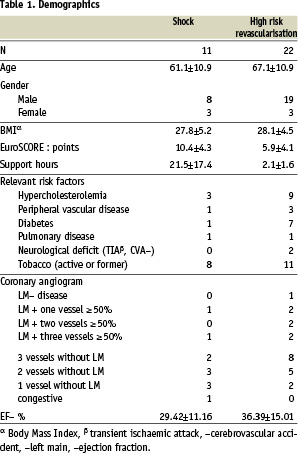
The indication for revascularisation is presented in table 2.
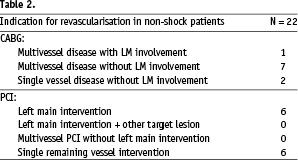
Overall these patients had left main or multiple vessel disease with poor left ventricle function. For group I the expected in-hospital mortality, based on shock trial registries is more than 50%. On the other hand, based on the EuroSCORE, the predicted mortality for the shock group is 9.78%,which might be an underestimation. For the elective revascularisation population (group II) based on the EuroSCORE, the predicted mortality is 3.89%.
The mean support time in group I was 21.5±17.4 hours (range 1.9 to 53.5 hours) and in group II 2.1 hours±1.6 (range 0.5 to 5.3 hours), indicating that none of these patients required further support after the PCI or OPCAB procedure.
Safety
Only 1 patient experienced limb ischaemia, 1 patient (2.8%, 95% CI: 0.07%, 15%) suffered from a nervus femoralis injury and 3 patients (8.3%, 95% CI: 1.8%, 22%) had bleeding complications (Table 3).
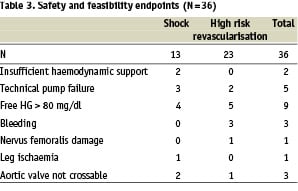
With the initial design a high number of patients had a rise of free haemoglobin of more than 80 mg/dl (n=9) (Table 4).
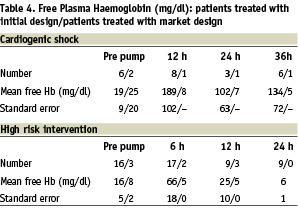
For this reason the stiffness of the cannula was increased to avoid kinking (kinking results in less performance and more haemolysis) and the distance between the impeller vanes and the pump housing was increased 0.04 mm thus reducing shear-stress by 100 N/m2. With this final design used in the last 5 patients no haemolysis above 80 mg/dl was observed (maximum = 46 mg/dl).
Shock patients were at higher risk to develop haemolysis, 4 of 13 patients > 80 mg/dl versus 5 of 23 patients in the elective group.
The free plasma haemoglobin increases quickly during the first 6 hours, thereafter a decline is observed.
Feasibility
In the first two patients the pump could not be placed over the aortic valve. As a consequence, a 0.014” wire was placed in the left ventricle and the pump was advanced in an over-the-wire technique. Since then this problem has not recurred. In one patient the pump was ejected early and could not be repositioned. The implantation procedure was graded easy in 23, suitable in 6, difficult in 3 and failed in 3 (non-reported in 1).
In total 5 technical pump failures occurred: in 3 patients the explantation was complicated by a fracture of the cannula with the pump housing remaining at the access site; in one patient the remaining part was retrieved surgically.
In 2 patients malfunction and early removal of the device occurred: in 1 patient this was due to a leak in the seal resulting in blood entering the electronic part of the pump; in the other it was due to a blockage of the impeller caused by atheromic plaque.
The pump remained in a stable position in most of the patients, and only small repositioning corrections were performed during follow-up.
Efficacy
The pump gave sufficient support in most of the patients. It should be mentioned, however, that in cardiogenic shock patients the pump was combined with an IABP (9/11). Even with both devices in place, due to haemodynamic instability, 2 patients crossed over to a device with more force (Recover® LP 5.0 and Medos®).
The haemodynamic data are presented in table 5.
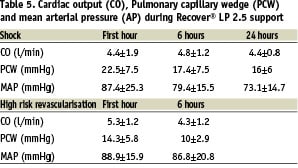
A small but significant increase, from 4.4±1.9 L/min to 4.8±1.2 L/min, in cardiac output was seen in the shock group (p=0.0178).
The major effect was, however, the decrease in filling pressures, most obvious in the cardiogenic shock group (PCW from 22.5±7.5 mmHg to 17.4±7.5 mmHg, p=0.00008), but still significant in the high-risk revascularisation group (PCW from 14.3±5.8 mmHg to 10±2.9 mmHg, p=0.0327). The mean arterial pressure did not change significantly (p=0.3772) in the shock group and slightly decreased in the high risk revascularisation group (–2.1 mmHg, p=0.0701). However, the decrease in pulsatility illustrates the relative importance of the pump flow32. Figure 4 shows the increase in motor current and decrease in pulsatility with a stable mean aortic blood pressure during a high-risk intervention (balloon inflation in the unprotected left main coronary artery).
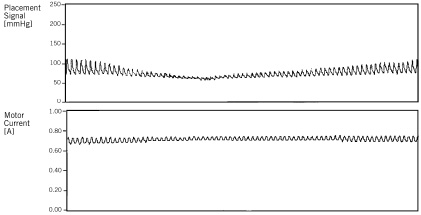
Figure 4. Amplitude of the motor current signal. Aortic pressure amplitude decreases during PCI.
At 6 months follow-up in the high-risk intervention group 4/22 patients died, of which one patient was 5 days post implantation due to an acute myocardial infarction. During the PCI procedure no major adverse cardiac events occurred. The other 3 patients died at a mean of 114 days post-procedure (range 82-159 days) indicative of the poor shape of the patients treated. Of the shock patients 6/11 supported patients died, 2 during support, and all within the first 30 days.
Discussion
We performed a phase I non-randomised multicentre trial to assess the safety, feasibility and efficacy of a new percutaneous left ventricle assist device. Previously the Recover® LP 2.5 was tested in animals via the carotid artery. Infarct size reduction, even with partial support, could be demonstrated32.
In this clinical trial, using the femoral approach, several issues had to be addressed and solved.
Firstly, during the trial a dedicated 13F peal away sheath and tapered access closure sheet was developed. As a consequence the catheter size in the femoral artery is reduced to 9F, reducing the risk of limb ischaemia and bleeding.
Secondly, an ‘over-the-wire’ technique was introduced. Initially, investigators, trying to advance the cannula across the aortic valve directly, encountered problems of kinking of the cannula. This kinking resulted in failure of crossing the aortic valve (in 2 patients). Since the introduction of this over-the-wire technique, no more failures were encountered.
The third important modification was the extension of the cannula with a pigtail which allows more aligned positioning of the cannula in the left ventricle and reduces the risk of cannula displacement.
In the application of high speed rotary blood pumps, haemolysis is an important concern. Haemolysis depends on the applied shear stress, which is assumed to be high, and the contact time, which assumed to be low34. It is also known that shock patients have a reduced free haemoglobin clearance due to poor liver function35. This explains why in the series haemolysis was more pronounced in the shock group. We observed an early peak in free haemoglobin at 6 hours with a sharp decline afterwards. We assume that this phenomenon is explained by the rapid haemolysis in pre-damaged and fragile cells. During the study the distance between the 2 vanes of the impeller and the pump housing was increased to reduce the shear forces. Due to the previously described (monorail technique, pigtail, increase space between vanes and pump housing) modifications, the last design, used in 5 patients, did not cause haemolysis. Free haemoglobin remained below 80 mg/dl. However, especially in patients with multiple organ failure, one should carefully monitor the free haemoglobin levels. A larger population needs to be studied with the final design to confirm the data.
In general, activation of the pump results in a decrease of the systolic-diastolic amplitude and an increase in the mean arterial pressure. Filling pressures are reduced substantially. The indication for usage of the Recover® LP 2.5 pump will remain high-risk revascularisations and patients presenting with haemodynamic instability, even though randomised data supporting the use of an assist device during high-risk interventions are scarce.
One should note that none of our patients died during the high-risk revascularisation procedure. The mortality rate at six months is indicative of the type of patients included in the protocol.
The effect on cardiac output in both groups was limited but significant. Patients in the cardiogenic shock group were on inotropes, and had a balloon pump on top, factors confounding the effect on cardiac output measurements.
It should be stated, however, that in patients with very low output, the small Recover® LP 2.5 pump will not provide full support and haemodynamic stability, therefore IABP was combined with Impella. In these patients more forceful assist devices should be used. Although in shock patients, the use of IABP has become common, data of randomised trials are lacking and the effect on mortality of the IABP as a stand-alone procedure is not clear19,20,22,23,25. On more advanced assist devices even less data is available28-30.
The most obvious effect of the pump was a clear reduction in preload of the left ventricle, as well in shock patients where the effect was most pronounced, as in patients who were haemodynamically stable but with increased LV-pressures secondary to their compromised LV-function. This unloading of the left ventricle might be beneficial in patients presenting with large acute myocardial infarction, resulting in infarct size reduction. Although animal data are promising32,33,35,36, data in humans are lacking at present.
Conclusions
Modifications of the initially designed pump led to a final new design which is a fairly easy to use and successful percutaneous left ventricle assist device with low rates of haemolysis. During high-risk revascularisation procedures haemodynamic stability was obtained. For cardiogenic shock patients it can be used as a tool for initial stabilisation, mainly reducing filling pressures and followed, if needed, by more aggressive treatment modalities.
Acknowledgements
We thank Mrs. H. Bollen for the excellent preparation of the article.
