Abstract
Aims: This study assessed safety and efficacy of a third-generation distal protection device, MedNova CardioShield Bare Wire Myocardial Protection System, for treating Saphenous Vein Graft (SVG) disease.
Treatment of SVG disease remains difficult, with increased adverse cardiac events (MACE) primarily manifested as no reflow and periprocedural infarction. Even with approved embolic protection devices, 30-day MACE rates are approximately 10%.
Methods and results: A multicentre randomized clinical trial evaluated a third-generation distal protection device MedNova CardioShield vs Percusurge GuardWire in 652 patients undergoing treatment of SVG disease, using a primary endpoint of 30-day death, Q-wave, non-Q-wave infarction, or target vessel revascularisation (MACE).
The primary endpoint occurred in 11.4% with CardioShield vs 9.1% with GuardWire (P=.37). Intention-to-treat analysis showed a strong trend for noninferiority (P=.057). Secondary modified intention-to-treat analysis including only patients receiving treatment device and no protocol deviation (defined as treatment of another lesion not using embolic protection) supported noninferiority of CardioShield (P=.022).
Conclusion: Analysis of outcomes of treatment strategies for SVG disease is difficult. In this trial, final results depended on whether a patient actually received the device according to protocol. With 30-day MACE as primary endpoint, CardioShield was not demonstrated to be noninferior to GuardWire.
Abbreviations
CAPTIVE = CardioShield Application Protects during Transluminal Intervention of Vein grafts by reducing Emboli
CK = creatine kinase
ECG = electrocardiography
EPD = embolic protection device
FDA = US Food and Drug Administration
FIRE = FilterWire EX Randomised Evaluation
MACE = major adverse cardiac events
MB = myocardial band
MI = myocardial infarction
OTW = over-the-wire
PCI = percutaneous coronary intervention
RX = rapid exchange
SAFER = Saphenous Vein Graft Angioplasty Free of Emboli Randomised
SVG = saphenous vein graft
TIMI = Thrombolysis in Myocardial Infarction
TVR = target vessel revascularisation
Introduction
The catheter-based treatment of a diseased Saphenous Vein Graft (SVG) is problematic because periprocedural adverse events occur with substantial frequency. The distal embolisation of the friable and often thrombus-containing degenerated SVG atheroma during Percutaneous Coronary Intervention (PCI) has been postulated as the mechanism for the increased incidence of Major Adverse Cardiac Events (MACE) associated with SVG treatment, primarily manifested as no reflow and periprocedural Myocardial Infarction (MI)1. Periprocedural increase of Creatine Kinase (CK) (Myocardial Band [MB] subunit) 5 times normal has been shown to be an independent predictor of cumulative morbidity and mortality2. Various strategies have been tested to decrease the incidence of MACE attributed to SVG treatment. These include conventional percutaneous transluminal coronary angioplasty, directional atherectomy, thrombectomy, ultrasound thrombolysis, excimer laser, stent implantation, and adjunctive treatment with glycoprotein IIB/IIIA inhibitors2-10. Until the advent of Embolic Protection Devices (EPDs), only stent implantation had been shown to significantly improve outcomes in SVG PCI3,4.
After satisfactory demonstration of safety and efficacy in decreasing MACE, 2 EPDs have been approved for use in the treatment of a diseased SVG. The Saphenous Vein Graft Angioplasty Free of Emboli Randomised (SAFER) trial demonstrated superiority of the GuardWire system (Medtronic, Minneapolis, Minnesota) to no protection during conventional SVG stenting5. The FilterWire EX (Boston Scientific, Natick, Massachusetts) was approved after the FilterWire EX Randomised Evaluation (FIRE) trial demonstrated noninferiority to the GuardWire system in a similar patient population6. Although the success of these 2 trials and availability of the devices have improved the safety of SVG intervention, the composite end point of 30-day MACE for protected SVG intervention is still approximately 10%.
Neither of the current devices is ideal, either because of the need to occlude distal flow (GuardWire) or the potential for incomplete capture of debris and movement of the filter within the SVG during stent placement and subsequent device exchanges. Accordingly, new devices are being designed and tested. A multicentre randomised trial, CardioShield Application Protects During Transluminal Intervention of Vein Grafts by Reducing Emboli (CAPTIVE) trial, tested the safety and efficacy of the MedNova CardioShield Vascular Protection System (MedNova, Galway, Ireland) for the treatment of patients with a single diseased SVG and the intention to treat the index lesion or lesions with stenting, with or without concurrent use of a glycoprotein IIb/IIIa inhibitor.
Methods
Patients with ischaemia due to stenotic SVG disease who met inclusion criteria were considered for enrolment in the CAPTIVE trial. Patients were randomly assigned to receive either the CardioShield (MedNova, Galway, Ireland) or GuardWire distal protection devices. Illustrations of CardioShield and GuardWire devices are shown in Figure 1.
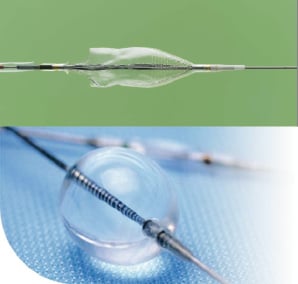
Figure 1. Investigational CardioShield embolic protection device deployed on a 0.014” Barewire guidewire (top), and the GuardWire Temporary Occlusion Balloon embolic protection device inflated on the system guidewire (bottom).
This randomised study complied with the Declaration of Helsinki regarding investigation in humans and was approved by the United States Food and Drug Administration (FDA). All 85 investigational sites (see Appendix) received approval from their hospital institutional review boards, and informed consent was obtained from all patients.
Patient selection
Patients were eligible for enrolment if they were 18 years of age or older and had myocardial ischaemia caused by a stenotic target lesion > 50% within a coronary artery bypass SVG. Clinical exclusion criteria included MI within the past 24 hours (with persistently elevated CK levels at the time of treatment), cerebrovascular accident within 2 months, planned treatment of another (non index) coronary stenosis, or left ventricular ejection fraction < 30%. Angiographic exclusions included an index lesion within 5 mm of the ostium, reference vessel diameter < 3.0 mm or > 6.0 mm, Thrombolysis in Myocardial Infarction (TIMI) flow grade < 2, lesion within 3.6 cm of the distal anastomosis for the 3.0 mm and 4.0 mm devices or within 4.3 cm for the 5.0 mm and 6.0 mm devices, or lesions located beyond the first anastomosis in jump grafts.
Randomisation and interventional protocol
After a patient gave consent and all inclusion criteria were achieved, randomisation in a 1:1 ratio was performed using block randomisation envelopes. Randomisation was stratified on the basis of the investigator’s intention to treat or not to treat with a glycoprotein IIb/IIIa antagonist. Aspirin 325 mg and clopidogrel 75 mg were administered before the procedure. Intravenous heparin was administered to maintain the procedural activated clotting time to > 300 seconds.
The CardioShield Bare Wire Myocardial Protection System is available as either an Over-The-Wire (OTW) or Rapid eXchange (RX) device. Both devices consist of a filtration element, delivery catheter, filter delivery wire, and retrieval catheter. The filtration element, consisting of a polyurethane outer membrane with internal supporting nitinol arms, is available in 3.0 mm, 4.0 mm, 5.0 mm, and 6.0 mm nominal diameters. The delivery catheter has a crossing profile of 0.046 inch to 0.051 inch for the OTW system and 0.047 inch to 0.051 inch for the RX system, depending on the filtration element size. The RX delivery catheter has a guidewire exit port 32 cm proximal to the distal tip. The 0.014 inch filter delivery wire has a 3-cm 0.018 distal radiopaque tip to retain the filter on the wire and to permit retrieval of the filter into the retrieval catheter. The filtration element is loaded into a pod located at the distal tip of the delivery catheter, advanced over the filter delivery wire across the lesion, and deployed by retracting the outer sheath of the delivery catheter. PCI is then performed over the filter delivery wire, with antegrade perfusion carrying liberated debris into the filter. On completion of PCI, the filter is retrieved into the expansile tip at the distal end of the retrieval catheter.
The GuardWire consists of a 0.014 inch guide wire with a central lumen to which an elastomeric balloon is attached distally5,7. The crossing profile is 0.036 inch. Once the balloon is distal to the lesion, it is inflated with diluted contrast, resulting in vessel occlusion. The syringe is then removed after sealing the central lumen with the MicroSeal adaptor, and PCI is performed over the wire with any 0.014 inch compatible device. Liberated debris is aspirated through the 5F monorail Export catheter, the balloon is deflated, and antegrade flow is restored.
Stenting was subsequently performed via standard techniques. Predilation to allow EPD delivery was discouraged. Although operators were allowed to predilate at their discretion, the majority of index lesions were treated with the use of a direct stenting technique. Blood samples for determining CK and CK-MB levels were collected at the start of each procedure and every 8 hours after the procedure at least 3 times and followed until the levels decreased. ElectroCardioGraphy (ECG) was performed before the procedure, immediately after the procedure, and before hospital discharge. Post-procedure, patients were treated with aspirin indefinitely, and clopidogrel was recommended for at least 30 days if a stent was placed.
Definitions, end points, and statistical analysis
The primary end point of the study was the 30-day composite of MACE, defined as death, Q-wave or non-Q-wave MI, or target vessel revascularisation, including emergent bypass surgery of the target vessel. The primary 30-day end point included in-hospital MACE. MI was defined as a Q-wave MI by the development of new, pathologic Q waves in 2 or more contiguous leads (assessed by a blinded ECG core laboratory) with a post procedure increase in CK or CK-MB levels above normal. Non-Q-wave MI was defined as an increase in post procedure CK-MB levels to > 3 times normal in the absence of new pathologic Q waves. Technical success for the CardioShield was defined as successful delivery and deployment of the device to the target site, device retrieval, and the presence of an intact filter at the end of the procedure. Technical success for the GuardWire was defined as delivery, inflation, and occlusion throughout the procedure without failure and aspiration, deflation, and removal without failure. Procedure success was defined as technical success with a final lesion diameter stenosis < 50% without in-hospital MACE. Angiographic analysis was performed in a central core laboratory with validated techniques8. A degeneration score was used to define the extent of disease. This score used the axial length of either luminal irregularities or ectasia to calculate the percentage of the entire SVG length involved with disease.
The study was designed to demonstrate that the CardioShield was noninferior to the GuardWire11. Assuming 30-day MACE rates of 10% for both the control and study device, it would be necessary to randomise 618 patients to demonstrate noninferiority with 80% power, a 1-sided α error of 5%, and δ of 6%. Categorical variables were compared by the likelihood ratio χ2 test or the Fisher exact test. Continuous variables were compared by 1-way ANOVA or unpaired t test. Independent clinical and angiographic predictors of 30-day MACE were determined using logistic regression. All P values are 2-sided with a statistical significance criterion of P<.05. The primary analysis included all patients based on the intention to treat. After randomisation, 28 patients (4% of all patients: 17 in the CardioShield arm and 11 in the GuardWire arm) were removed from the analysis population because either the device was never deployed (n=21 subjects) or a protocol-prohibited unprotected intervention occurred before or immediately after treatment (n=7). A secondary modified intention-to-treat analysis was performed after excluding these patients.
Results
Patient population
Between December 2001 and January 2004, 652 patients at 85 sites in the United States and Europe undergoing stent implantation were randomly assigned to the 2 groups. Baseline clinical characteristics were well matched between the 2 groups, with no important differences in the frequency of cardiac risk factors (Table 1).
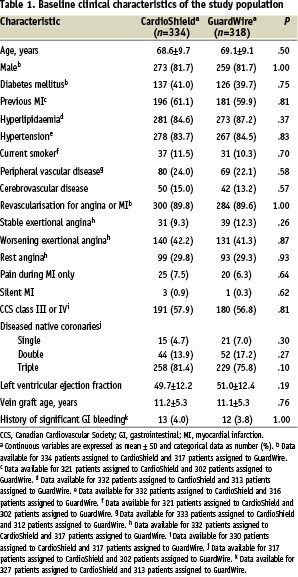
Comorbid conditions were frequent, with peripheral vascular disease in 24.0% and 22.1% of the CardioShield group and GuardWire group, respectively, cerebrovascular disease in 15.0% and 13.2%, and worsening angina in 42.2% and 41.3%. The mean age of the SVG was 11.2 years for the CardioShield group and 11.1 years for the GuardWire group. The baseline characteristics of the vein grafts were also similar between the 2 groups (Table 2).
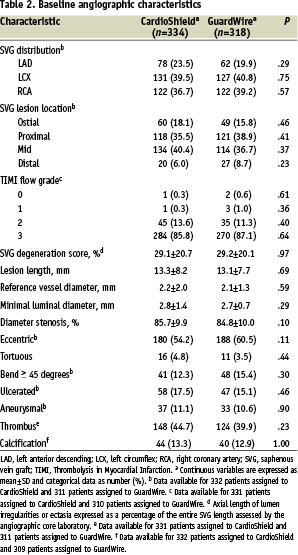
Approximately 40% of vein grafts contained thrombus, and the extent of disease contained within the vein graft in both arms was comparable (~30%).
Procedural performance and results
A glycoprotein IIb/IIIa inhibitor was used at the operator’s discretion in approximately 40% of patients overall (Table 3).
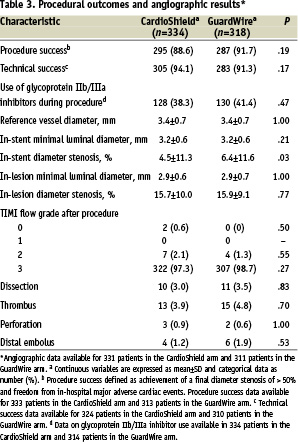
Procedural and technical success rates were high at approximately 90%. The final diameter in-stent stenosis in the CardioShield group was slightly smaller than in the GuardWire group (4.5% vs 6.4%, P=.03). At the end of the procedure, final TIMI 3 flow rates were excellent in both groups, at 97.3% and 98.7% for the CardioShield and GuardWire groups, respectively.
In-hospital and 30-day clinical outcomes
The primary end point (30-day MACE rate) of the intention-to-treat study was 11.4% for the CardioShield arm and 9.1% for the GuardWire arm (P=.37) (Table 4).
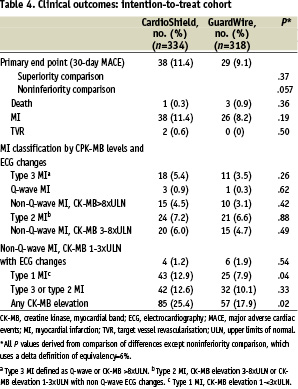
In-hospital MACE occurred in 11.4% of the CardioShield group compared with 8.8% of the GuardWire group (difference [95% CI]= 2.6% [–20 to 7.2], P=.30). As shown in Figure 2, the composite MACE was driven by non-Q-wave MIs.
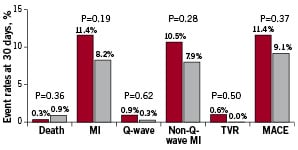
Figure 2. Intent-to-treat analysis. Thirty-day major adverse cardiac events (MACE) rates in the CardioShield (n=334) (black bars) and GuardWire (n=318) (white bars) groups. MI, myocardial infarction; TVR, target vessel revascularisation.
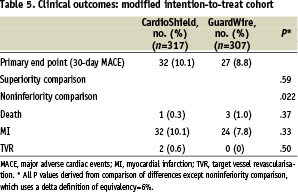
Although the overall rate of MI was statistically similar in the 2 groups (non-Q-wave and Q-wave), the rate of type 1 MI was higher in the CardioShield group (Table 4). Although no statistically significant difference between the rates of the primary end point was found between the 2 treatment arms, the intention-to-treat analysis failed to support the alternative hypothesis of noninferiority (P=.057). As mentioned above, 28 patients (4% of the total group) either had failure of the attempt to deliver the randomised treatment assignment or had a protocol deviation, with treatment of another unprotected lesion. This group was excluded and a secondary modified intention-to-treat analysis was performed. The results of this analysis supported the noninferiority of the CardioShield device (P=.022) (Table 5).
In the univariate analysis of MACE at 30 days stratified by baseline clinical and angiographic characteristics, the CardioShield and GuardWire rates were similar except in lesions with a higher plaque volume, for which the MACE rates were lower in the GuardWire arm (Table 6).
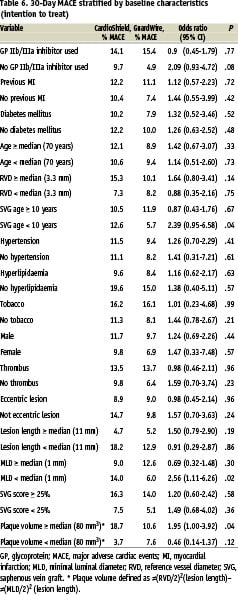
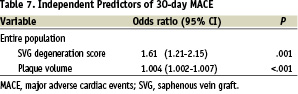
In the multivariable model, the only independent predictors of 30-day MACE were increasing plaque volume and higher SVG degeneration score (Table 7).
Discussion
Under a strict intent-to-treat analysis, the current trial, designed to assess the safety and efficacy of the CardioShield filter-based EPD, showed a strong trend but failed to demonstrate noninferiority to the FDA-approved control balloon-occlusion EPD, GuardWire. The protocol pre-specified P value (false-positive rate) that was needed to demonstrate noninferiority was .05, but the final test for noninferiority yielded a P value of.057. Under a modified intent-to-treat analysis, performed post hoc with the objective to remove subjects (n=28 of 618 patients randomised) who did not receive either assigned therapy or who had additional coronary or vein graft lesions treated without EPD (thus confounding the potential source of cardiac enzyme elevations), the test for noninferiority was positive (P=.022) (Table 5).
The incidence of 30-day MACE in the CardioShield intention-to-treat cohort, 11.4%, and in the modified intention-to-treat cohort, 10.1%, is lower than the historical control rates of MACE in previous studies of SVG PCI, which have ranged from 15% to 30%2,12, including the most recent evaluation of SVG PCI without distal protection in the control arm of the SAFER trial, with a 30-day MACE rate of 16.5%5. In the FIRE trial, which resulted in FDA approval of the FilterWire EX device, the 1-sided test of noninferiority was met and prospective users could anticipate relative 30-day adverse event rates that were from 45% better to 33% worse than those of the GuardWire6.
In the present study, the rate of Type 1 MI was significantly higher in the CardioShield group (12.9% vs 7.9%, P=.04). The prognostic significance of low level increases in CK-MB periprocedurally has been a matter of considerable debate. Whereas the efficacy of EPDs is predicated on their ability to capture debris liberated during PCI (this has been demonstrated by analyses of filters or aspirate retrieved after PCI13-15), it is possible that technical limitations of the device, suboptimal lesion selection, or suboptimal deployment by the operator or a combination of these factors could account for the differences in 30-day MACE observed in the trial16. The significantly higher degree of low level CK-MB elevation might suggest a technical limitation of the CardioShield relative to the GuardWire; alternatively, greater experience with the GuardWire could also have exaggerated a difference in device performance or efficacy.
The current treatment of vein graft disease is not optimal, with periprocedural in-hospital adverse events, predominantly non-Q-wave MI, occurring in approximately 10% of patients even with the benefit of an EPD. The concept of distal protection is intuitively attractive. That a MACE rate of approximately 9% to 11%, predominantly non-Q-wave MIs, remains regardless of the protection device used is of concern. Potential mitigating factors include incomplete capture of debris from suboptimal device occlusion or sizing, release of neurohumoral or vasoactive factors at PCI, embolisation during initial device passage, or embolisation of plaque debris from the stented segment or elsewhere within the SVG after the protection device has been removed. One advantage of filter-based devices is the ability to leave the devices in the SVG for longer periods after stenting, provided satisfactory flow is maintained. This longer period of protection should be considered, especially when TIMI flow grade 2 or better is present with the filter deployed. Options such as leaving the filter device in for several additional minutes after stenting or gentle lavage therapy within the stented segment (containing extruded soft, friable atheroma) and through the filter may effectively reduce the incidence of periprocedural non-Q-wave MI. New approaches and strategies, including proximal protection, need to be investigated and eventually optimised to improve the safety of SVG PCI.
Conclusions
This randomised trial of a new third-generation distal protection device showed a strong trend but failed to support the hypothesis of noninferiority using an intention-to-treat analysis plan compared with GuardWire; the prespecified P value was .05, but the final analysis yielded a P value of .057. In a second modified intention-to-treat analysis that excluded patients who did not receive either assigned therapy or who had a protocol violation with additional lesions treated without EPD, the test for noninferiority was positive (P=.022). In a multivariable model, the only independent predictors of 30-day MACE were increasing plaque volume and high SVG degeneration scores.
Acknowledgment
Editing, proofreading, and reference verification were provided by the Section of Scientific Publications, Mayo Clinic.
Appendix
Study sites: Affiliated Cardiologists of Arizona, Phoenix, AZ; Amphia Hospital, Breda, the Netherlands; Andreus Greuntzig Haus, Hamburg, Germany; Atlanta Heart and Vascular Group, Atlanta, GA; AZ Middleheim Hospital, Antwerp, Belgium; Baptist Hospital of East Tennessee, Knoxville, TN; Baptist Montclair Hospital, Birmingham, AL; Barts and the London Chest NHS Hospital Trust, London, UK; Bridgeport Hospital, Bridgeport, CT; Buffalo General Hospital, Buffalo, NY; Cardiology Consultants, Pensacola, FL; The Caregroup, Indianapolis, IN; Christiana Care Health Services, Newark, DE; The Cleveland Clinic Foundation, Cleveland, OH; Clinique Pasteur, Toulouse, France; Covenant Medical Center Cardiovascular Research, Saginaw, MI; Dartmouth Hitchcock Medical Center, Lebanon, NH; Dayton Heart Center, Dayton, OH; Drs. Baker and Gilmour, Jacksonville, FL; Emory Healthcare Heart Center, Atlanta, GA; Evanston Northwestern Healthcare, Evanston, IL; Florida Cardiovascular Research/JFK Medical Center, Atlantis, FL; Harrisburg Hospital, Harrisburg, PA; Hartford Hospital, Hartford, CT; The Heart Center, Huntsville, AL; Henry Ford Hospital, Detroit, MI; Hesperia Hospital, Modena, Italy; Hoag Hospital, Macon, GA; Institut Cardiovasculaire Paris Sud, Institut Jacques-Cartier, Massy, France; King’s College Hospital NHS Trust, London, UK; Lahey Clinic, Burlington, MA; Lenox Hill Hospital, New York, NY; Mater Misericordiae Hospital, Dublin, Ireland; Mayo Clinic Arizona, Phoenix, AZ; Medical Group of Fort Wayne Northern Indiana Research Alliance, Fort Wayne, IN; Memphis Heart Clinic, Memphis, TN; Mercer University School of Medicine, Newport Beach, CA; Mid America Cardiology at KU Medical Center, Kansas City, MO; Mid America Heart Institute, Kansas City, MO; Mid-Ohio Cardiology and Vascular Consultants, Columbus, OH; North Ohio Heart, Cleveland, OH; North Ridge Heart Institute, Ft. Lauderdale, FL; North Shore University Hospital, Manhasset, NY; Northeast Cardiology Associates, Bangor, ME; Northern California Research Institute, San Francisco, CA; NYU Medical Center, New York, NY; Pacific Foundation for Cardiology Research, Redwood City, CA; Phoenix Heart Center, Phoenix, AZ; Providence Hospital, Southfield, MI; Rockford Cardiology Associates, Rockford, IL; Rush Presbyterian Hospital, Chicago, IL; St. Antonius Hospital, Nieuwegein, the Netherlands; St. Francis Hospital, Roslyn, NY; St. James’s Hospital, Dublin, Ireland; St. Joseph’s/Phoenix, Phoenix, AZ; St. Marys Hospital, Rochester, MN; Scripps Memorial Hospital, LaJolla, CA; Seattle Cardiovascular Research, Bellevue, WA; Sentara Norfolk General Hospital, Norfolk, VA; Shawnee Mission, Shawnee Mission, KS; South Carolina Heart Center, Columbia, SC; Spokane Cardiology, Spokane, WA; Strong Memorial Hospital, University of Rochester, Rochester, NY; Temple University Hospital, Philadelphia, PA; Thomas Jefferson Medical Center, Philadelphia, PA; Universitätsklinikum Essen, Essen, Germany; University Hospital Rotterdam, Rotterdam, the Netherlands; University of Alabama, Birmingham, AL; University of Arizona Heart Center, Tucson, AZ; University of Chicago, Chicago, IL; University of Colorado Health Sciences Center, Denver, CO; University of Florida, Gainesville, FL; University of Iowa, Iowa City, IA; University of Massachusetts Memorial Medical Center, Worcester, MA; University of Michigan Hospital, Ann Arbor, MI; University of North Carolina, Chapel Hill, NC; University of Pittsburgh Medical Center Presbyterian, Pittsburgh, PA; University of Texas Health Sciences Center, San Antonio, TX; University of Texas Medical School, Houston, TX; UZ Gasthuisberg Leuven, Leuven, Belgium; Virginia Cardiovascular Specialists, Richmond, VA; Washington Hospital Center, Washington, DC; West Side Cardiology Associates, Cleveland, OH; Westchester Medical Center, Valhalla, NY; York Hospital, York, PA.