Abstract
Background: Everolimus has been successfully tested in humans using both an erodable and a durable polymer in small previous studies.
Methods: This single blind multi-centre non-inferiority randomised (3:1) controlled trial evaluated the safety and performance of the XIENCE V Everolimus Eluting Coronary Stent System (XIENCE V EECSS) versus the TAXUS Paclitaxel Eluting Coronary Stent System (TAXUS® PECSS) in the treatment of patients with a maximum of two de novo native coronary artery lesions located in two different epicardial vessels. Three hundred patients with evidence of myocardial ischaemia were allocated to stent implantation with an everolimus-eluting stent (n=223) or a paclitaxel-eluting stent (n=77). Suitable lesions had a diameter stenosis of <50-99%, a length of <28 mm, and a reference vessel diameter between 2.5 mm and 4.25 mm. The primary endpoint was in-stent late loss (LL) at 180 days. Percentage in-stent volume obstruction (%VO) was measured by intravascular ultrasound (IVUS) in a subset of 152 patients. Clinical secondary endpoints included ischaemia driven major adverse cardiac events (ID-MACE) at 180 days.
Results: At 6 months, the in-stent LL was 0.11±0.27 mm in the everolimus-eluting stent arm, as compared to 0.36±0.39 mm in the paclitaxel-eluting stent arm (p<0.0001). Percentage VO in the everolimus-eluting stent arm was 2.5±4.7% versus 7.4±7.0% in the paclitaxel-eluting stent arm (p<0.0001). Hierarchical MACE was 2.7% (6/222) in the everolimus-eluting stent arm vs. 6.5% (5/77) in the paclitaxel-eluting stent arm.
Conclusion: This non-inferiority randomised trial not only met its primary endpoint, but also demonstrated the superiority of the everolimus-eluting stent over the paclitaxel-eluting stent in terms of in-stent late loss.
Introduction
Recent studies that have evaluated the local application of anti-proliferative drugs (sirolimus and paclitaxel) for the prevention of restenosis via a stent delivery system have shown that these therapies successfully inhibit the development of neointimal hyperplasia and reduce restenosis and associated clinical events.1,2
Everolimus is an effective anti-proliferative agent that inhibits growth factor-stimulated cell proliferation by causing cell cycle arrest in the late G1 stage in the cell cycle.3
The feasibility of using everolimus on a drug-eluting stent was demonstrated in the earlier FUTURE I4,5 and FUTURE II6,7 studies and more recently in the SPIRIT FIRST8 study, using the everolimus-eluting stent. The SPIRIT FIRST study (N=60) was a multi-centre, single blinded controlled study conducted to assess the feasibility and efficacy of the everolimus-eluting stent in the treatment of patients with de novo native coronary artery lesions compared to the metallic, uncoated MULTI-LINK VISION RX Coronary Stent. This feasibility trial showed clinical safety and the angiographic in-stent Late Loss (LL) observed was 0.10 mm, a reduction of 88% relative to the bare metal stent at six months and an in-stent LL of 0.24 mm at 12 months, which was a reduction of 71%.8,9
The SPIRIT II trial is a continuation of the assessment of the safety and performance of the XIENCE V everolimus-eluting stent versus the TAXUS paclitaxel-eluting stent in the treatment of patients with a maximum of two de novo native coronary artery lesions.
Methods
Patient selection
This prospective, randomised (3:1) single-blind, parallel two-arm trial was performed at 28 centres in Europe, India and New Zealand and enrolled patients from July 2005 to November 2005. It was approved by the ethics committee at each participating institution, and all patients gave written informed consent.
Patients were eligible for the study if they were older than 18 years and had evidence of myocardial ischaemia. The patient could have a maximum of two de novo native coronary artery lesions, which had to be located in different major epicardial vessels. The de novo target lesion(s) had to have a reference vessel diameter between 2.5 mm and 4.25 mm by visual estimation, a target lesion length <28 mm, a visually estimated stenosis between 50-99% of the luminal diameter, and a Thrombolysis In Myocardial Infarction (TIMI) flow grade of 1 or more. Patients were not eligible for enrolment if they had known diagnosis of acute myocardial infarction three days prior to the baseline procedure, a left ventricular ejection fraction of less than 30%, were awaiting a heart transplant, or had a known hypersensitivity or contraindication to aspirin, heparin, bivalirudin, clopidogrel or ticlopidine, cobalt, chromium, nickel, tungsten, everolimus, paclitaxel, acrylic and fluoro polymers or contrast sensitivity that could not be adequately pre-medicated. Additionally, patients having target lesion(s) with an aorto-ostial or left main location, a lesion located within 2 mm of the origin of the left anterior descending- or left circumflex, heavy calcification, or a visible thrombus within the target vessel were also excluded from the trial.
The everolimus-eluting stent
The XIENCE V Everolimus Eluting Coronary Stent System (EECSS) (Advanced Cardiovascular Systems, an Abbott Vascular Company, IL, USA) is comprised of the ACS MULTI-LINK VISION Stent and delivery system, and a drug eluting coating. The ACS MULTI-LINK VISION Stent is a balloon expandable stent, which consists of serpentine rings connected by links fabricated from a single piece of medical grade L-605 cobalt chromium alloy.
Everolimus is blended in a non-erodable polymer, coated over another non-erodable polymer primer layer. The coating comprises acrylic and fluoro polymers, both approved for use in blood contacting applications. This layer of everolimus-polymer matrix with a thickness of 5-6 microns is applied to the surface of the stent and is loaded with 100 micrograms of everolimus per square centimetre of stent surface area with no topcoat polymer layer. The stent is designed to release approximately 80% of the drug within 30 days after implantation.
Everolimus (Certican®, Novartis Corporation) has been evaluated in clinical trials in the US and Europe for use as an immunosuppressant following cardiac and renal transplantation.10 Everolimus has received market approval in the European Union and the XIENCE V EECSS has received CE mark in the European Union.
Study procedure
Following the confirmation of angiographic inclusion and exclusion criteria prior to the procedure, patients were enrolled through a telephone randomisation service and assigned in a 3:1 ratio to either an everolimus-eluting stent or a paclitaxel-eluting stent. The stents were available in lengths of 8, 18 and 28 mm, and diameters of 2.5, 3.0, 3.5 and 4.0 mm. Lesion lengths greater than 22 or less than or equal to 28 mm were to be covered by 2 stents; twice an 18 mm stent, or a 28 mm and an 8 mm stent.
Lesions were treated using standard interventional techniques with mandatory pre-dilatation and stent implantation at a pressure not exceeding the burst pressure rate. Due to packaging differences, physicians were not blinded to the device. Post-dilatation was left to the discretion of the physician, however, if performed, it was only to be done with balloons sized to fit within the boundaries of the stent. In the event of a bailout procedure and additional stent requirement, the stent had to be one from the same arm as the first implanted stent. IVUS was performed in a subset of 152 consecutive patients enrolled in pre-selected centres, after angiographically optimal stent placement had been obtained, and was repeated if additional post-dilatation was performed to optimise stent apposition and/or deployment.
Peri-procedural pharmaceutical treatment was administrated according to standard hospital practice. Either unfractionated heparin or bivalirudin could be used for procedural anticoagulation. The use of glycoprotein IIb/IIIa inhibitors was left to the discretion of the physician. All patients enrolled into the study were pre-treated with a loading dose of 300 mg of clopidogrel and maintained on 75 mg of clopidogrel daily for a minimum of 6 months and >75 mg of aspirin daily for a minimum of one year following the procedure.
Clinical device success was defined as a successful delivery and deployment of the first inserted study stent (in overlapping stent setting a successful delivery and deployment of the first and second study stent) at the intended target lesion with attainment of final residual stenosis of 50% of the target lesion by QCA (by visual estimation if QCA unavailable). Bailout patients were included as clinical device success only if the above criteria for clinical device success were met.
Clinical procedure success included the previous criteria of clinical device success, but with the addition of any study stent or other stent devices and required the absence of ID-MACE during the hospital stay. In dual lesion setting both lesions had to meet clinical procedure success.
Follow-up
Patients were evaluated at 30 and 180 days. Further evaluations will be performed at 270 days, 1 and 2 year(s) and will form the subject of additional reports. At outpatient visits, patients were asked specific questions about the interim development of angina or the occurrence of MACE. Angiographic follow-up for all patients and IVUS in a subset of 152 consecutive patients (enrolled at selected centres) were performed at 180 days, and both investigations will be repeated at 2 years for this subset of patients. Prior to performing a follow-up angiogram, the physician was required to record prospectively in the eCRF whether a revascularisation (if required) was clinically indicated – defined as the presence of ischaemic symptoms and/or a positive functional ischaemia study.
Quantitative coronary angiography evaluation
Quantitative coronary angiography was performed using the CAAS II analysis system (Pie Medical BV, Maastricht, Netherlands).11 In each patient, the stented segment and the peri-stent segments (defined by a length of 5 mm proximal and distal to the stent edge) were analysed. The following QCA parameters were computed: minimal luminal diameter (MLD), reference vessel diameter (RVD) obtained by an interpolated method, and percentage diameter stenosis (%DS). Binary restenosis (BR) was defined in every segment as diameter stenosis >50% at follow-up. Late loss (LL) was defined as the difference between MLD post-procedure and MLD at follow-up.
Intravascular ultrasound analysis
Post-procedure and follow-up stented vessel segments were examined with mechanical or phased array intravascular ultrasound (Eagle-eye™ Volcano, Atlantis™, Boston Scientific) using automated pull-back at 0.5 mm per second. The coronary segment beginning 5 mm distal to and extending 5 mm proximal to the stented segment was examined. A computer-based contour detection program was used for automated 3-D reconstruction of the stented and adjacent segments. The lumen, stent boundaries and external elastic membrane (vessel boundaries) were detected using a minimum cost algorithm.12 The stent volume (SV) and lumen volume (LV) were calculated according to the Simpson’s rule.13 The intrastent neointimal volume was calculated as the difference between SV and LV. The percentage obstruction of the stent volume was calculated as intrastent neointimal volume/stent volume*100. Feasibility, reproducibility and inter- and intra-observer variability of this system have been validated in vitro and in vivo.13 Incomplete apposition was defined as one or more stent struts separated from the vessel wall with evidence of blood speckles behind the strut on ultrasound, while late-acquired incomplete apposition was defined as incomplete apposition of the stent at follow-up which was not present post-procedure.14-16
Study endpoints
The primary endpoint was angiographic in-stent LL, as determined by quantitative angiography, based on an “analysis lesion”: one randomly selected lesion per patient to avoid inter-lesion dependence20.
Secondary endpoints (QCA and IVUS) at 180 days and 2 years (subset of 152 consecutive patients enrolled at selected centres) included the in-stent, in-segment, proximal and distal LL; in-stent and in-segment angiographic binary restenosis rate and %DS; in-stent percentage volume obstruction (%VO) and plaque behind the stent; and persisting and late-acquired incomplete stent apposition, aneurysm, thrombosis and persisting dissection. In-stent was defined as within the margins of the stent while in-segment was defined as located within the margins of the stent and 5 mm proximal or distal to the stent. Late loss was calculated as the difference between the post-procedure and follow-up minimum luminal diameters.
Secondary clinical endpoints included Ischaemia-Driven MACE (comprised of cardiac death, myocardial infarction and Ischaemia-Driven Target Lesion Revascularisation [ID-TLR]) either by CABG or PCI, evaluated at 30, 180 and 270 days, 1 and 2 year(s) after the index procedure and acute success including clinical device and clinical procedure success.
All deaths that could not be clearly attributed to another cause were considered cardiac deaths.
A non-Q-wave myocardial infarction was defined as a typical rise and fall of CK-MB* with at least one of the following: ischaemic symptoms, ECG changes indicative of ischaemia (ST segment elevation or depression) or coronary artery intervention. (*if non-procedural/spontaneous MI, CK-MB >2 times upper limit of normal; if post PCI, CK-MB >3 times upper limit of normal; if post CABG, CK-MB >5 times upper limit of normal).
ID-TLR was defined as a revascularisation at the target lesion associated with any of the following: non-invasive positive functional ischaemia study (e.g. exercise testing or equivalent tests) or invasive positive functional ischaemia study (e.g. Fractional Flow Reserve [FFR] or Coronary Flow Reserve [CFR]); ischaemic symptoms and an angiographic%DS >50% by on-line quantitative coronary angiography (QCA);%DS >70% by on-line QCA without either ischaemic symptoms or a positive functional study. The investigator assessment could potentially be overruled by QCA off-line from the core laboratory.
Stent thrombosis, categorised as acute (<1 day), subacute (> 1 day <30 days) and late (> 30 days), was defined as any of the following: in the presence of angiography, clinical presentation of acute coronary syndrome17 with angiographic evidence of stent thrombosis. In the absence of angiography: cardiac death or acute MI in the territory of the stented vessel/vessels; AMI that could not be distinctly attributed to a non-target vessel during the Clinical Events Committee adjudication was considered in the composite for stent thrombosis.
The endpoints were adjudicated by an independent clinical events committee (appendix I). In addition, a data and safety monitoring board that was not affiliated with the study reviewed the data to identify any safety issues related to the conduct of the trial (appendix I).
Statistical analysis
The primary endpoint and all trial endpoints were analysed on both the intent-to-treat and per-treatment evaluable populations, the latter of which consisted of patients who had no major protocol deviations, as evaluated in a blinded manner.
The sample size for the study was determined based on the primary endpoint of in-stent LL at 180 days and on the following assumptions: one-tailed non-inferiority test, overall α equals 0.05, randomisation ratio was 3 (everolimus arm):1 (paclitaxel arm), the true mean in-stent late loss was assumed to be 0.32 mm in the XIENCE V arm and 0.39 mm in the TAXUS arm, a non-inferiority margin delta (δ) of 0.16 mm and the group sequential design was based on the method described in Reboussin, et al. indexed by O’Brien & Fleming boundary.18,19 Four interim analyses were planned and the final analysis was performed at the 0.0448 adjusted significance level. Given the above assumptions, analysing 180 patients in the test arm and 60 patients in the active control arm provides more than 91% power. In order to account for drop-outs and to ensure enough angiographic data, approximately 300 patients had to be enrolled of which 225 in the everolimus arm and 75 in the paclitaxel arm.
In this paper binary variables were compared using the Fisher’s exact test. Continuous variables were compared using Wilcoxon two-sample test. The hypothesis testing for the primary endpoint was performed using a one-sided non-inferiority test with asymptotic test statistic. If non-inferiority would be shown, superiority analysis was planned using a two-sided t-test at the 5% alpha level. Due to inclusion of dual vessel/lesion treatment, as a secondary analysis, a repeated measures analysis using all target lesions was performed and compared with the analysis using ‘analysis lesion’. Final 6-month results are presented in this manuscript.
Results
Patient characteristics
Between July 2005 and November 2005, 223 patients were randomly assigned to receive the everolimus eluting stent, and 77 were assigned to receive the paclitaxel eluting stent. As defined in the protocol, all results are presented for the intent-to-treat population; 222 patients in the everolimus arm, and 77 patients in the paclitaxel arm (Figure 1).
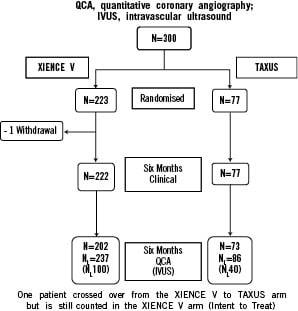
Figure 1. Flowchart of patients.
In the everolimus arm there was one withdrawal prior to 180 days. The two arms were similar with respect to baseline clinical variables examined in Table 1.
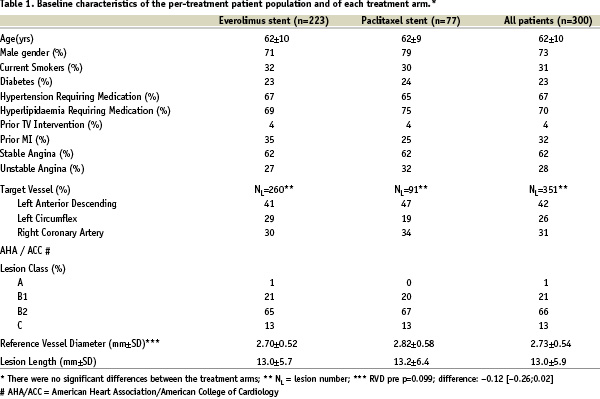
Procedural characteristics
The lesions in the two arms were treated similarly with the use of conventional techniques. Per patient, 1.4 study stents were implanted in the everolimus arm and 1.3 in the paclitaxel arm. Mean stent deployment pressure was 15 atmospheres in each arm and post dilatation was performed in 39% of lesions in the everolimus arm and 27% of lesions in the paclitaxel arm. Bail out study stents were used in 5.4% of lesions in the everolimus arm and 4.5% of lesions in the paclitaxel arm. Both arms had similar rates of clinical device success 98.8% (256/259) for the everolimus arm vs. 98.9% (89/90) for the paclitaxel arm and they did not differ significantly with respect to the rate of clinical procedure success 99.1% (221/223) in the everolimus arm and 97.4% (75/77) in the paclitaxel arm.
Quantitative coronary angiography analysis
Angiographic data at 180 days was available for 275 analysable patients (92%). Pre-procedure, the RVD of the everolimus arm tended to be smaller than in the paclitaxel arm without reaching statistical significance. Post-procedure, this difference became significant at 5% alpha. The significantly smaller MLD pre-procedure and a slightly smaller acute gain in the everolimus arm resulted in a statistically significant difference in post -procedure MLD (2.49mm vs. 2.62 mm; –0.13 [–0.24;–0.03]) (Table 2). At 180 days, the mean in-stent LL (analysis lesion, intent-to-treat population) was significantly lower for the everolimus arm compared to the paclitaxel arm, 0.11±0.27mm versus 0.36±0.39mm (non-inferiority p<0.0001, superiority p<0.0001). (Figure 2)
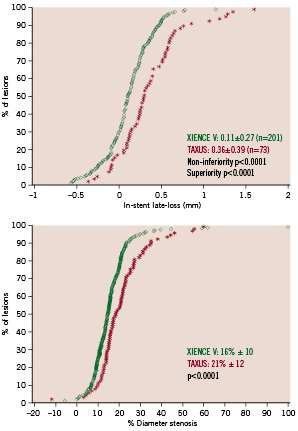
Figure 2. Cumulative frequency of in-stent late loss (analysis lesion) and in-stent percentage diameter stenosis at follow-up (all lesions).
For the per lesion analysis, the mean in-stent MLD,%DS and BR rate were 2.38±0.50 mm, 16±10% and 1.3% (3/237), respectively in the everolimus-eluting arm, as compared to 2.27±0.54 mm, 21±12%, and 3.5% (3/86) in the control arm. Figure 2 shows the cumulative distribution frequency curve of diameter stenosis at 180 days in each treatment arm. Table 2 shows the results of sub-segmental quantitative angiographic analysis for both treatment arms.
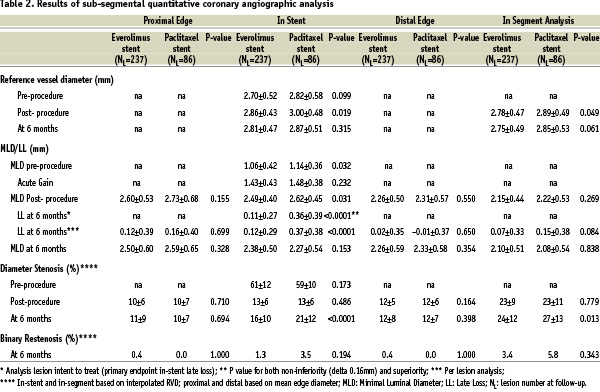
The in-segment, proximal, and distal LL were non-statistically different between the two arms. However, the in-segment %DS was significantly lower in the everolimus arm.
Intravascular ultrasound evaluation
At 180-days, intravascular ultrasound evaluation showed no significant differences between the two arms with respect to the volume of the stent or the lumen volume (Table 3).
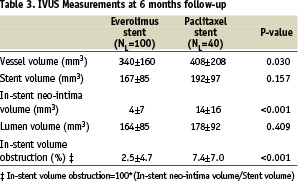
However, there was a significant difference in vessel volume which reflects the small imbalance in vessel size seen at baseline between the everolimus and paclitaxel arms and the nominal stent volume (calculation based on the nominal stent diameter and stent length) which was 186 mm3 in the paclitaxel arm and 173 mm3 in the everolimus
arm. Significantly less neointimal hyperplasia was observed in the everolimus-eluting stent arm compared to the paclitaxel-eluting stent arm (4±7 mm3 vs. 14±16 mm3, p<0.001) and similarly, significantly less %VO, (2.5±4.7% vs. 7.4±7.0%, p<0.001). Figure 3 shows the cumulative frequency distribution curve of %VO.
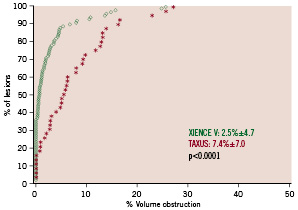
Figure 3. Cumulative curve of in-stent percentage volume obstruction.
Of the seven patients in the everolimus arm in which post-procedure stent malapposition was observed, three were persisting, three were resolved and one was not evaluable at 180 days. In the paclitaxel arm both cases of stent malapposition observed post-procedure were resolved at 180 days. There were no cases of late acquired stent malapposition in either arms.
Major adverse cardiac events
Major adverse cardiac events (MACE) are listed in Table 4.
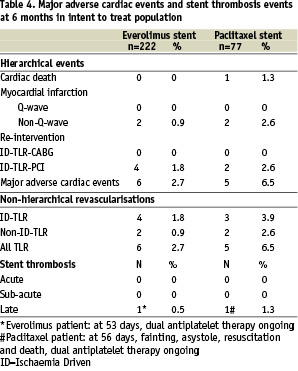
Hierarchically, for the intent-to-treat population in the everolimus arm two (0.9%) non-fatal non-Q wave MIs and four (1.8%) ID-TLRs by PCI were identified compared to one cardiac death (1.3%), two (2.6%) non-fatal non-Q wave MIs and two (2.6%) ID-TLRs by PCI in the paclitaxel arm. The total hierarchical MACE rate was 2.7% (6/222) in the everolimus-eluting arm vs. 6.5% (5/77) in the paclitaxel-eluting arm. In addition there were two (0.9%) ID-TVRs (non-target lesions) in the everolimus arm and none in the paclitaxel arm.
There were 0.9% (2/222) and 2.6% (2/77) non-ID TLRs in the two arms respectively.
There were no occurrences of acute or sub-acute stent thromboses in either arm. One case of late stent thrombosis occurred in the everolimus arm at 53 days following a complex procedure with multiple stent implants. One case of late stent thrombosis occurred also in the paclitaxel arm at 56 days post procedure. The latter patient presented with a myocardial infarction and subsequently died. Both patients were taking dual antiplatelet therapy at the time of their thrombotic event.
Discussion
The Spirit II trial has met its primary endpoint, namely it shows an in-stent late loss in the everolimus arm, which is not only non-inferior but also superior to the in-stent late loss observed in the paclitaxel arm.
At the time of the design of the trial, it was decided – in order to avoid potential inter-lesion dependence20 – to analyse only one lesion per patient (selected by a randomised process) for the primary endpoint, when the patient had received a stent in two different target vessel lesions (17% in the XIENCE V arm and 18% in the TAXUS arm). When all lesions were included in the analysis, the in-stent late loss remained unchanged (0.11 mm vs. 0.12 mm in the XIENCE V arm and 0.36 mm vs. 0.37 mm in the TAXUS arm). (Table 2).
Although a 3:1 randomisation everolimus vs. paclitaxel was performed, which provided more precision for the everolimus arm, without loss of power for the comparison; this might have resulted in a small imbalance in baseline characteristics pre- and post-procedure.
A trend towards a smaller pre-procedural vessel size in the everolimus arm was observed and this difference became significant post-procedure. These differences in vessel size and MLD post-procedure between the two arms could have impacted the restenosis rate and late loss as frequently demonstrated in the literature.21-23 The MLD post-procedure in the everolimus arm is significantly smaller than the MLD post-procedure in the paclitaxel arm; this is the result of a smaller pre-treatment MLD (p-value 0.032) combined with a smaller acute gain (ns), although the deployment was done at equal levels of pressure. Despite this potential handicap at baseline, in-stent LL and%DS at follow up were significantly lower in the everolimus arm. This small difference in vessel size at baseline is also exemplified in stent volumes measured at baseline (162 mm3 vs. 195 mm3) and at follow-up (167 mm3 vs. 192 mm3) between the everolimus and paclitaxel arms respectively. Although this difference in stent volume does not achieve significance, it could have also impacted the late proliferative process as previously reported in the literature.24 Nevertheless, we found a profound and highly significant reduction (73% reduction) in neointimal volume in the everolimus arm (3.8 mm3) when compared to the paclitaxel arm (14.4 mm3). Of interest was that in the IVUS findings of the SIRIUS study, assessing the efficacy of a DES coated with a comparable limus, an almost equal neointimal volume of 4.1 mm3 was found.
Whether malapposition can be held responsible for late stent thrombosis in patients who receive drug-eluting stent remains so far unknown.25,26 In the present population both drug-eluting stents show negative values of late loss, but the frequency of observations of negative late loss values within the everolimus arm is higher than in the paclitaxel arm (71/237=30% vs. 14/86=16%). The largest negative value was observed in the everolimus arm (–0.57 mm compared to –0.37 mm) in the paclitaxel arm. However, we must recognise that late loss is a parameter with a rather large standard deviation when inter-observer variability is assessed (1 SD 0.36 mm, 2 SD 0.72)11; and that late loss is the result of two individual measurements (MLD post-procedure, and MLD at follow-up) which both have their own inter-observer variability due mainly to the process of calibration.27 Therefore, a negative late loss of –0.57 mm is still within the limits of the confidence level for the reproducibility of the late loss parameter. To investigate the relationship between late loss and malapposition, we have examined the lesions (n=23) with negative late loss, which were assessed by IVUS at follow-up and which received an everolimus-eluting stent and which could therefore potentially have a late-acquired or persisting stent malapposition. Among the 23 lesions with a negative late loss, there was not a single case of late-acquired malapposition, and only one case of persisting malapposition.
In the present study, the incidence of diabetics in the everolimus- and paclitaxel arms was 23% (51/223) and 24% (18/76) respectively. The in-stent LL in the paclitaxel arm for the diabetic patients was 0.39 mm, which is comparable to the previously reported late loss of 0.43 mm in a meta-analysis of the diabetic subsets of the TAXUS family trials.28 In contrast, the LL in the diabetic patients in the everolimus arm was only 0.15 mm (SD 0.26) and thereby significantly superior to the loss of the paclitaxel arm which was 0.39 mm (SD 0.34). The difference in in-stent LL between everolimus- and paclitaxel-eluting stent was 0.24 mm (95% confidence interval: –0.41 mm; –0.08 mm). It is noteworthy that this difference is identical to the difference in late loss observed in the whole population, and thus indicates also superiority of everolimus-eluting stent versus the paclitaxel eluting stent in terms of LL reduction in the diabetic subset. However as this was not a pre-planned analysis further studies will be required to confirm this.
Conclusions
This non-inferiority randomised trial not only met its primary endpoint, but also demonstrated the superiority of the everolimus-eluting XIENCE V stent over the TAXUS paclitaxel-eluting stent in terms of in-stent late loss. In addition, the IVUS results showed that the XIENCE V stent was more effective at reducing neointimal hyperplasia than the TAXUS stent. The incidence of major adverse events was low and comparable between both treatment arms.
Acknowledgements
The authors would like to acknowledge the invaluable assistance of Cecile Dorange, MSc, and Susan Veldhof, RN, from Abbott Vascular, Diegem, Belgium for the conduct and reporting of this study and for their assistance in the preparation of this manuscript.
Appendix I
Sponsor: Advanced Cardiovascular Systems, an Abbott Vascular Company, Santa Clara, California, USA.
Principal investigator: Patrick W. Serruys (The Netherlands).
Steering committee: Patrick W. Serruys (Principal Investigator and Chairman, Rotterdam, The Netherlands); Gary Johnson (Vice President of Regulatory Affairs, Pre-Clinical Research, Clinical Research and Reimbursement, Cardiac Therapies, Abbott Vascular); Marcus Wiemer, Bad Oeynhausen, Germany; Eulogio Garcia, Madrid, Spain; John Ormiston, Auckland, New Zealand.
Data Safety Monitoring Board (DSMB): Jan G.P. Tijssen, Amsterdam, The Netherlands; Thierry Lefèvre, Massy, France; Philip Urban, Meyrin-Geneva, Switzerland.
Clinical Events Committee (CEC): Claude Hanet, Clinique Universitaire de Saint-Luc, Brussels, Belgium; Dougal McClean, Christchurch, Christchurch, New Zealand, Victor Umans, Medical Center Alkmaar, The Netherlands.
Data Management – Angiographic and IVUS Core Laboratories: Cardialysis BV, Rotterdam, The Netherlands;
Data Coordination Centre and Site Monitoring: Advanced Cardiovascular Systems, and Abbott Vascular Company, Diegem, Belgium.
The following investigators and institutions participated in the SPIRIT II Trial:
J.J. Piek, Academisch Medisch Centrum, Amsterdam, The Netherlands; P. Ruygrok, Green Lane Cardiovascular Service, Auckland, New Zealand; J. Neuzner, Klinikum Kassel, Kassel, Germany; A. Seth, Max Heart and Vascular Institute, New Delhi, India; J. Schofer, Universitäres Herz-und Gefässzentrum Hamburg, Hamburg, Germany; M. Wiemer, Herzzentrum Bad Oeynhausen, Bad Oeynhausen, Germany; G. Richardt, Segeberger Kliniken GmbH, Bad Segeberg, Germany; D. Carrié, Hôpital de Rangueil CHU, Toulouse, France; L. Thuesen, Skejby Sygehus, Aarhus, Denmark; E. Camenzind, R.V. Hôpital Cantonal Universitaire de Genève, Geneva, Switzerland; H. Kelbaek, Rigshospitalet, Copenhagen, Denmark; P.W. Serruys, Erasmus Medical Center, Rotterdam, The Netherlands; C. Macaya, Hospital Clinico San Carlos, Madrid, Spain; J. Berland, Clinique Saint Hilaire, Rouen, France; M. Desaga, Amper Kliniken AG-Klinikum Dachau, Dachau, Germany; F. Van den Branden, A.Z. Middelheim, Antwerpen, Belgium; K. Rasmussen, Aalborg Sygehus Syd, Aalborg, Denmark; H. Suryapranata, Isala Kliniken Locatie Weezenlanden, Zwolle, The Netherlands; V. Legrand, C.H.U. de Liège Sart Tilman, Liège, Belgium; W. Ruzyllo, National Institute of Cardiology in Warsaw, Warsaw, Poland; A. Manari, Azienda Ospedaliera Santa Maria Nuova, Reggio Emilia, Italy; C. Spaulding, Hôpital Cochin, Paris, France; M. Suttorp, St. Antonius Ziekenhuis Nieuwegein, Nieuwegein, The Netherlands; J. Boland, C.H.R. La Citadelle, Liège, Belgium; K. Huber, Wilhelminenspital der Stadt Wien, Vienna, Austria; E. Garcia, University Hospital Gregorio Maranon, Madrid, Spain; J.A.M. te Riele, Amphia Hospital, Breda, The Netherlands; P. Ruygrok, The Mercy Hospital, Auckland, New Zealand.
