Abstract
Atherosclerosis is currently appreciated as a disease with a large inflammatory component. The underlying mechanisms, which are uncovered in a rapid pace, are greatly interconnected and as such very complex. Nevertheless, for clinicians it is important have some degree of insight in these immunologic mechanisms in order to interpret the current research advances. The aim of this review is to supply clinicians with this knowledge, avoiding too much detail. All the relevant immunologic basics will be discussed at first, followed by the immunity related theories of atherosclerosis. Finally, current and new immune-modulatory therapies will be discussed.
Abbreviations list
ACE: Angiotensin converting enzyme
APC: Antigen presenting cell
ApoE: Apolipoprotein type E
DC: Dendritic cell
eNOS: Endothelial nitric oxide synthase
HDL: High density lipoprotein
HMG CoA: 3-hydroxy-3-methylglutaryl coenzyme A
HSP: Heat shock protein
IFN-γ: Interferon-gamma
IL: Interleukin
LDL: Low density lipoprotein
LPS: Lipopolysaccharide
MCP-1: Monocyte chemotactic protein-1
MDA: Malondialdehyde
MHC: Major histocompatibility complex
MMP: Matrix metalloproteinase
Mφ: Macrophage
NO: Nitric oxide
OxLDL: oxidised low density lipoprotein
SR: Scavenger receptor
TLR: Toll like receptor
TNF-α: Tumor necrosis factor alpha
VALT: Vascular associated lymphoid tissue
Introduction
Atherosclerosis remains the disease with the highest mortality in the Western world. In the United States 47% of overall cardiovascular mortality is related to this disease. While for many years atherosclerosis has been regarded as a lipid-driven disease, it is now clear that the immune system critically contributes to this pathogenic process1. Lipid metabolism and inflammation mutually influence each other yielding the complete spectrum of atherosclerotic disease progression. This is reflected in the strong correlation between future cardiovascular events and the combined values of cholesterol, as indicator of lipid status, and CRP, as indicator of systemic inflammatory activity2. Since CRP levels reflect systemic inflammatory activity, this notion underscores the role of immunity in human atherosclerosis.
The first part of this review concisely introduces the immune system basics most relevant for atherosclerosis. In the second part, an overview is given of the immune-related theories of atherosclerosis, while the third and final part discusses new immune-modulating therapies for atherosclerosis.
Inflammation driven by integrated innate and adaptive immune mechanisms
Although our awareness of the complexity of the immune system is expanding continuously, the principles of immune regulation of inflammation are increasingly well understood. This part of the review gives a current state of the art overview of these fundamentals. The figures and tables cover and summarise the contents of this part, while the references include more comprehensive reviews and detailed studies on the different subjects.
Figure 1 outlines the generic inflammatory cascade, which appears to be remarkably applicable to the events observed in the arterial wall upon initiation and progression of atherosclerotic lesions.
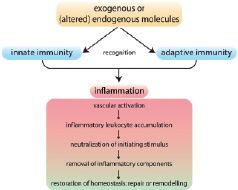
Figure 1. The inflammatory cascade in atherosclerosis, involving collaboration of innate and adaptive immunity, follows a common pathway. The inflammatory response is triggered by recognition of specific molecules by cells of innate and adaptive immunity that lead to their activation. The ensuing inflammatory cascade is initiated by lipid uptake in the vessel wall, which facilitates accumulation of leukocytes. These mobile defence forces attempt to neutralise and remove the initial trigger. If successful, inflammation subsides and homoeostasis can be restored by tissue repair. Semi-successful elimination triggers tissue remodelling, allowing partial functionality. Adapted from P.M. Henson108.
Central to the initiation of this cascade is the recognition of exogenous or (altered) endogenous molecules by cells of both innate and adaptive immunity. Activation of cells in both arms leads to an integrated response, triggering the pathogenic vascular effects characteristic of the disease. In the following paragraphs, we focus in particular on these early steps of recognition and the ensuing cellular response, and apply these to the specific conditions of atherosclerosis, since recent developments in this area have led to exciting new insights in the pathogenesis of inflammatory diseases, including atherosclerosis.
Innate versus adaptive immunity?
Table 1 globally compares innate with adaptive immunity.
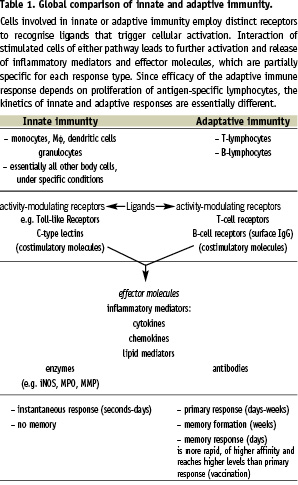
The principal difference between the two arms of the immune system is the nature of the cellular receptors involved in recognition of potentially pathogenic molecules and the triggering of these cells, which leads to cellular activation. Receptors of adaptive immunity, expressed only by T- and B-lymphocytes, are generated by recombination of gene segments, while receptors of innate immunity are encoded as such within the genome, not requiring genetic recombination. Leukocytes involved in innate immunity such as monocytes, macrophages (Mφ), dendritic cells (DC) and granulocytes express a large variety of receptors recognising molecules of pathogens. For example, they can express receptors for bacterial compounds such as lipopolysaccharide (LPS) and for viral RNA (see also Tables 2 and 3, for receptors and ligands, respectively).


The specificity of these receptors is fixed by the nature of their static genetic encoding. In general, all body cells can make use of several innate immune mechanisms involving such receptors. However, the leukocytes of innate immunity are unique in the fact that they provide a mobile response and are specialised in expressing various host defence mechanisms upon triggering. As a consequence of their mobility and functional specialisation, these cells can rapidly accumulate at affected tissue sites in case of injury or infection and create an effective response.
In contrast to innate immunity, adaptive immunity is exclusively restricted to T- and B-lymphocytes. These cells employ intricate mechanisms to rearrange gene building blocks, thus constructing novel receptors. Such receptors are cell-specific (clonotypic) and the expressing cells are subsequently selected for optimal recognition of an antigenic ligand and clonal expansion. The T-cell receptor expressed on the surface of the T lymphocyte specifically recognises pathogenic or self-peptides presented by major histocompatibility (MHC) class II (for the CD4+ T helper cells) or MHC class I (for the CD8+ cytotoxic T-cells). MHC molecules are the scaffolds expressed by antigen presenting cells (APC), which expose peptides to lymphocytes, controlling responses in infection and transplantation. B-lymphocytes use their unique cell surface immunoglobulin as antigen-specific receptor and, after activation, secrete these as antibodies that are able to recognise epitopes on intact proteins.
The innate response to potentially harmful substances provides immediate protection (seconds to hours), while the adaptive response takes longer to develop (days), but has the unique properties of very high specificity and memory formation, with a stronger and higher affinity response upon second exposure (i.e. the principle of vaccination). Although the definition of adaptive versus innate immunity is clear-cut (yes or no genetic recombination to form receptors), these two arms of the immune system are completely integrated and intertwined, since they depend on mutual activation for an optimal response (see middle part of Table 1). Moreover, T and B-lymphocytes express several innate receptors in addition to their cell-specific antigen receptor. Upon activation by recognition of cognate ligands, cells of both innate and adaptive immunity produce soluble signalling molecules, including cytokines and chemokines. In addition, they express sets of costimulatory molecules on their surface guiding cellular interactions and modulating the ultimate response to the initial pathogenic stimulus. So, although the definition of the two separate arms is clear, innate and adaptive immunity work in coordination and jointly. Much of this coordination and integration is brought about by APC, in particular Mφ and DC. The cells of the Mφ/DC lineage are specialists in immune regulation by virtue of their high level expression of the relevant surface molecules and the ability to migrate and thus convey environmental information to and from the adaptive immune system. This environmental information is particularly sensed by a variety of innate antigen receptors that have been discovered over the last years and which appear to belong to large families of partially unknown molecules (Table 2).
Atherosclerosis: innate and adaptive immunity out of control?
The role of the immune system in atherosclerosis has gained acceptance in the last decade because of a series of experiments performed mainly by human pathologists in parallel to experimental vascular biologists. Pathologists showed that in addition to foamy Mφ, also T-cells, B-cells and DC are present in human atherosclerotic plaques3-6. Since MHC class-II and costimulatory molecules like CD40, CD80 and CD86 were also found to be expressed in plaques obtained at autopsy7-11, important requirements for local activation of adaptive immune mechanisms are fulfilled. Recent evidence suggests that activated T-cells in both human and mouse plaques are of oligoclonal origin12,13, implying selective activation by a limited number of antigenic epitopes. The expression of Toll-like receptors (TLR) in human plaques, especially in vulnerable plaques, allows the activation of the innate immune system not only by microbial ligands, but also by potential endogenous ligands such as modified LDL and heat shock proteins14,15 (Figure 2).
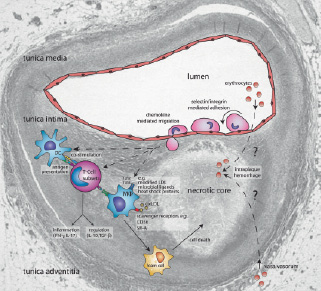
Figure 2. Immune mechanisms in atherosclerosis projected onto a vulnerable plaque section. This schematic view displays selected mechanisms discussed in the review, with special emphasis on immune modulatory therapy. The histological section displays a lumen-narrowing plaque, which consists of a large necrotic core, covered by a thin fibrous cap. This is a typical example of a vulnerable plaque. In brief, monocytes and lymphocytes roll over, and adhere to the dysfunctional vascular endothelium, and subsequently migrate into the vascular wall. Next, monocytes will differentiate into dendritic cells and macrophages. Macrophage uptake of oxLDL results in the formation of foam cells, which form a major constituent of growing plaque. Demise of foam cells by necrosis or apoptosis contributes to formation of the necrotic core. Activation of T-cells by macrophages and dendritic cells results in the differentiation of T-cells into distinct subsets that either promote inflammation (Th1, Th2, Th-IL-17) or limit the inflammatory process (Treg). Also, the neovascularisation in the plaque and its vasa vasorum is displayed, which possibly leads to intraplaque hemorrhages. Please note that the cartoon components are not drawn to scale with respect each other, nor to the underlying section. This plaque section was previously published in 109. Adapted with permission from The American College of Cardiology Foundation.
These human studies were paralleled by studies in experimental atherosclerosis models, in particular focusing on chemokines and cytokines, both soluble mediators of inflammation. Chemokines, a family of proteins that control the migration of inflammatory cells play a dominant role in atherogenesis16. Inhibition of various chemokines reduces plaque formation to different degrees16-23. In addition, production of pro-inflammatory cytokines by activated immune cells has a pro-atherogenic effect, while the anti-inflammatory cytokine IL-10 limits atherosclerosis24-27. Inhibition of pro-inflammatory compounds clearly mediates a reduction in atherosclerosis28-30.
These findings on the role of the immune system in atherosclerosis have all been excellently and extensively reviewed recently, with emphasis on general aspects31, innate immunity31-35, adaptive immunity31 and immunomodulation36.
The role of innate immunity in atherosclerosis
The monocyte-macrophage lineage is considered to play a central role in innate immunity as well as in atherosclerosis. Studies in mice deficient for Chemokine (C-C motif) receptor 2 (CCR2), the monocyte receptor which mediates transmigration into the vessel wall by MCP-1, show a reduction in atherosclerosis, indicating that monocyte-derived cells, presumably (activated) Mφ aggravate atherosclerosis. Although studies in MCP-1 deficient mice further confirmed these early findings, recent studies indicate that the MCP1-CCR2 axis is particularly important during early atherosclerosis and that additional stimuli are necessary for advanced plaque formation37 (see below).
An important way of macrophage stimulation is by TLR ligation, in particular TLR4 and TLR2, (recently reviewed in reference 38)38. Lack of TLR-signalling in pro-atherogenic backgrounds of the mouse gives rise to decreased disease severity. In line with the mouse studies, some epidemiological data suggest that altered TLR function, caused by gene polymorphisms, protects from atherosclerosis (discussed in reference 39)39. This might be due to changed ligation of TLR by pathogens and their derived products like LPS. Hence, these receptors have been mentioned as a connecting factor between atherosclerosis and other parameters, including circulating plasma endotoxins, Chlamydia pneumoniae infections and periodontal disease (Porphyromonas gingivalis). Taken together, these findings suggest that the fast and efficient reactions against bacterial pathogens may increase progression of atherosclerosis.
The importance of the innate immune system in atherosclerosis is also demonstrated by the ubiquitous presence in the plaques of the macrophage scavenger receptors, which include CD36, SR-B1, SR-A, and less known receptors such as FEEL-1, SR-PSOX and CD163 (Table 2). The scavenger receptors are low affinity receptors with broad ligand specificity, which play a role in the uptake of oxidised low density lipoprotein, oxLDL (CD36, SR-B1, SR-A and SR-PSOX) and the uptake of hemoglobin (CD163). They lack feedback inhibition of activity by intracellular cholesterol levels and as such load macrophages with oxLDL and play a key role in foam cell formation, one of the hallmarks of atherogenesis.
In conclusion, the innate system is capable of rapid activation of the monocyte-macrophage cell lineage through a variety of mechanisms, thereby contributing to atherosclerosis by driving proinflammatory mechanisms. This has led to a theory of repeated activation of innate immunity in atherogenesis by some authors35,40,41.
The role of adaptive immunity in atherosclerosis
It is clear from the above-mentioned studies that the immune system plays a dominant role in atherogenesis. In addition to innate mechanisms, adaptive immunity also plays a role in atherogenesis. Important components of adaptive responses are expressed in plaques, including MHC class II, costimulatory molecules and Th1 cytokine profiles31,35. Many studies, mainly performed in ApoE or LDL-R deficient mice, show that inhibition of a specific part of adaptive immunity inhibits atherogenesis. Ligands relevant in the activation of adaptive immunity are listed in Table 3.
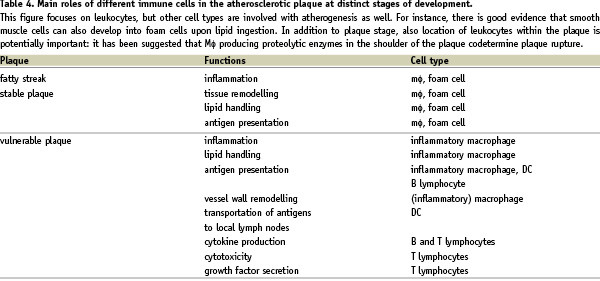
More recently, the view has emerged that adaptive immunity may be particularly important in the regulation of plaque phenotype; i.e., stable vs. vulnerable plaque, which may rupture and lead to myocardial or cerebral infarction42. Vulnerable plaques display a specific cellular composition43, including many inflammatory cells and little stabilising connective tissue. It is presently unknown how this plaque composition develops, but inhibition of (certain components of) adaptive immunity has been shown to induce a decrease in atherosclerosis44,45. Therefore, a major conclusion from this research is, that the activation of adaptive immunity is associated with the severity of atherosclerosis (Table 4).
As adaptive immunity needs an antigenic trigger to become activated, recent studies focused on the identification of specific antigens and epitopes involved in atherogenesis. Several candidates were put forward: oxLDL, Chlamydia, endogenous HSP60 and microbial HSP65 (Table 3). Especially the first candidate, oxLDL, is of importance as it may be regarded as a modified self-protein. Consequently, it has been suggested that atherosclerosis is an autoimmune disease35. Currently, the discussion is lively and of great importance as identification of such antigens and epitopes in principle allows for development of vaccines against atherosclerosis. Such a vaccine should induce immune responses generating protective antibodies or, alternatively, long-lasting immune tolerance. However, approximately 100 different epitopes stimulating adaptive immunity have been identified in the oxLDL moiety, which renders vaccine development very difficult.
Is atherosclerosis an (auto-)immune disease?
A straightforward answer to the question whether atherosclerosis is an immune disease in origin is dependent on definitions in play. As mentioned above, lipid metabolism and inflammation is functionally closely interconnected and involvement of both systems is a prerequisite for atherosclerotic disease to develop. The abundant presence of leukocytes with effector functions operational in the atherosclerotic plaque leaves little room for denying the involvement of immunity. Could atherosclerosis be mediated by autoimmunity, an idea much supported by work from Wick and colleagues35,46. By definition, autoimmunity is mediated by T- and B-lymphocytes with adaptive receptors for self antigen. Indeed, evidence has been found for antibodies against endogenous HSP and for both antibodies and T-lymphocytes reactive against the self-compound LDL modified by oxidation. However, it is exceedingly difficult to prove that self-reactivity is pathogenically relevant, since self-reactivity has been demonstrated in healthy individuals as well. The importance is obvious, since vaccination strategies are being developed, which require identification of pivotal auto-antigens involved (see above). Studies in mouse models, although limited by species differences and relatively high lipid doses used, collectively indicate that there is evidence that immune mechanisms are responsible for atherosclerosis21,31,35,47-50. For instance, T- and B-lymphocytes stimulate lesion initiation or progression49. Under lower atherogenic pressure, lymphocytes do affect lesion development, in particular plaque vulnerability. Furthermore, cytokines promoting and limiting inflammation similarly promote and limit atherosclerosis.
Do infectious agents critically influence atherosclerosis? Several studies have identified Chlamydia pneumoniae as a major suspect, but convincing evidence in this respect is lacking and clinical trials using antibiotics have not demonstrated a protective effect51. Other microbial suspects have also been suggested, but with similar lack of conclusive evidence. Two intriguing concepts with respect to microbe involvement in atherogenesis warrant further investigation, namely the possible pathogenic effects of multiple infections with different pathogens (the pathogen burden hypothesis41) and contributions of the normal healthy gut and other mucosa bacterial flora to vascular inflammation41. In conclusion, atherosclerosis cannot be considered an auto-immune disease as it is not a solely immune driven disease, evidenced by plaques in severe combined immune deficient mice45, but merely as a disease with auto-immune like characteristics.
Promising research issues on immunity and atherosclerosis
As summarised in Table 5, many questions remain unanswered and drive future research into pathogenesis and therapy.

For instance, what is the role of vascular-associated lymphoid tissue (VALT) in plaque localisation and initiation? After pioneering work by Wick et al the fascinating hypothesis that localisation of atherosclerosis is related to local VALT in the vessel wall has not been studied in depth by other laboratories, while it may explain several observations35, like the predilection sites of atherosclerosis and the occurrence of atherosclerosis in the arterial system and not in the venous system. Furthermore, new subsets of monocytes in the blood and activation levels of Mφ in the plaque have been identified that may shed some light on inflammation control within plaques5,52,53. Another important observation is the strict upstream location of inflammatory components in plaques54. This indicates a spatially oriented control mechanism, perhaps due to local interaction between cytokines, chemokines, leukocytes and endothelium, which might be related to the local differences in haemodynamic parameters55,56. In addition, recently several new animal models for vulnerable plaque have been developed, of which the brachiocephalic trunk model by the group of Jackson57 and the low shear stress mouse model of Cheng et al both strongly indicate a role of blood flow in vulnerable plaque initiation and progression57-59. Finally, an important new field is the therapeutic immunomodulation of atherosclerosis, discussed in the next paragraph.
Immunomodulatory therapies of vulnerable plaque
Several therapies directly or indirectly affect the immune system and thereby plaque progression and vulnerable plaque formation. After a small introduction focused upon classical treatment and its effect on the immune system, an overview is given on newer treatment modalities specifically related to the inflammatory component of atherosclerosis (Table 6).
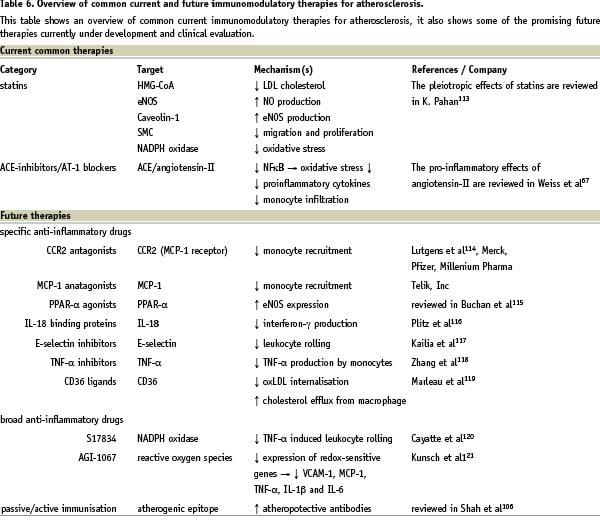
Classical interventions
Experimental studies suggested that angiotensin converting enzyme (ACE) inhibitors might have an anti-atherosclerotic effect, inhibiting plaque progression and even stabilising plaque composition, mostly by a reduction in angiotensin-II levels60-63. This results, for example, in reduced MCP-1 production and decreased foam cell formation. Angiotensin-II inhibition has also been shown to lower matrix metalloproteinase (MMP) levels, which are particularly produced by activated macrophages64. The production of these enzymes has a central role in the pathophysiology of vessel remodeling65. The added beneficial effect of angiotensin-II receptor type I (AT-I) blockers on myocardial infarction compared to ACE-inhibitors is still ambiguous66. For further reading, the anti-inflammatory effects of ACE-inhibitors67 and the pro-atherogenic effects of angiotensin-II were reviewed previously68. More recently, human studies on carotid arteries demonstrated the therapeutically beneficial effect of ACE inhibitors on atherosclerosis progression in vivo69,70. In conclusion, the effects of ACE-inhibitors on atherosclerosis are much broader than just decreasing systemic risk factors like hypertension and therefore deserve a role in treatment regimens.
One of the current cornerstones in treatment of coronary arterial disease are HMG-CoA reductase inhibitors (statins). They restore endothelial synthesis of nitric oxide (NO) in presence of hyperlipidaemia, which is crucial to preserve endothelial function. Thus, beneficial effects of statins are not limited to LDL reduction71-78, as they also increase expression of eNOS79 and interfere with superoxide formation80,81. Statins have been shown to increase eNOS expression by various mechanisms, such as restoring the L-arginine transport82, which is the substrate for eNOS, and by decreasing caveolin-1, which blocks the interaction between eNOS and its cofactor (calcium/calmodulin)83.
It has been hypothesised that the cardiovascular risk reduction by statins may be due to changes in both plaque composition and its pleiotropic biological effects on its components, rather than a simple reduction in plaque size84-86. These pleiotropic effects may include a reduction in the expression of MHC class II, a shift in T-cell balance from Th1 towards Th2 and reduced leukocyte adhesion to the endothelium87. Experiments have also shown that statins have a potent antiproliferative effect on smooth muscle cells88. Additionally, simvastatin appears to reduce neointimal hyperplasia, which is the primary cause of restenosis after stent placement. Also, it enhances re-endothelialisation, which protects against in-stent thrombosis.
Furthermore, regression of atherosclerotic plaques in the peripheral circulation by intensive statin therapy has been reported89-92. The mechanisms are still not fully elucidated, but changes in LDL and HDL cholesterol levels are described90,93,94.
Vulnerable plaques are reported to have an increased extracellular matrix breakdown, resulting in a (localised) weak spot of the plaque. This is mediated by activity of proteases, which comprise several families: serine-proteases, cysteine-proteases and MMP95,96. MMP belong to a family of zinc-activated proteases modulating the extracellular matrix in the vascular wall. The activity of some family members induces weak spots in the extracellular matrix. MMP have been studied most extensively and many studies provided evidence for a role of these proteins in plaque vulnerability. Some studies showed an up-regulation of MMP in ruptured plaques compared to stable plaques95-98. In particular, overexpression of MMP-9 in Mφ is suggested to induce plaque rupture in otherwise stable plaques99. Apparently not all MMP negatively influence plaque development, since synthetic inhibitors of MMP, like tetracyclines and the macrolide Batimastat, have been shown to exert little effect on atherosclerotic tissue100. Therefore, MMP inhibition of specific family members is desirable for a MMP modulating therapy to be successful in atherosclerosis treatment.
Novel interventions
Since it is now generally accepted that activity of the immune system is affecting the initiation, progression and composition of plaques, several immunomodulatory therapies are proposed and tested in animal models. Manipulation of the immune system can, for example, be performed through either active or passive immunisation, both resulting in circulating neutralising antibodies, which may dampen the inflammatory response in atherosclerosis. Immunomodulatory therapies can be divided into those inhibiting the pro-atherogenic effects of the immune system or into those interventions stimulating the regulatory capabilities101.
Activation of TLR is, in general, thought to induce or enhance atherosclerosis by promoting inflammation. Indeed, several mouse lines employing inhibition of TLR4 showed a reduction in atherosclerotic plaques size38.
Compared to an innate immune response, an adaptive response takes longer to develop, but is more selective and can be functionally skewed into different directions. It needs an antigen to become activated and several antigens that may be involved in atherogenesis have been proposed in the literature (see Table 3). On the other hand, properly functioning immunity is atheroprotective mediated by naturally circulating IgM antibodies that are produced by B-lymphocytes. These antibodies attenuate murine atherosclerosis101. Two strategies have been reported that aimed at increasing the levels of these naturally occurring antibodies. Binder et al102 showed that immunisation of mice with malondialdehyde (MDA)-modified LDL, increased IgG1 antibodies to MDA-LDL and, surprisingly, also IgM antibodies to oxLDL102. Faria-Neto et al passively immunised mice using a monoclonal antibody against phosphorylcholine, an oxLDL headgroup, to study the effect on atherosclerosis103. They found that this treatment resulted in an increased titer of naturally occurring antibodies against the same epitope. Both experimental studies showed reduced atherosclerosis, although in the latter study the effect was only demonstrated in vein graft atherosclerosis. Interestingly, activation of the immune system through immunisation with oxLDL leads to a large reduction in atherosclerosis suggesting that this activation pathway has atheroprotective properties104-106. Another antigen to be considered in atherosclerosis is endogenous HSP60, which has cross-reactive epitopes with bacterial HSP60 and HSP65107. A bacterial infection could result in antibodies to bacterial HSP that may also interact with human HSP epitopes on stressed arterial cells. In contrast to using modified LDL epitopes as antigen, immunisation of LDLR –/– mice or hypercholesterolemic rabbits with HSP60 augments atherosclerosis104-106. Given the differential responsiveness of the immune system upon triggering with atherosclerosis-related molecules, i.e. stimulating either anti- or pro- atherogenic pathways, great care needs to be taken in the development of vaccine strategies aimed at the inhibition of atherosclerosis.
Acknowledgements
R. Krams is a recipient of the established investigator grant of the Dutch Heart Foundation (project 2002T045). C. Cheng was supported by the Interuniversity Cardiology Institute of the Netherlands (ICIN, project 33).
