Abstract
Aims: This paper reports the technical feasibility of using the Direct Flow Medical percutaneous aortic valve (PAV) to treat patients with severe aortic stenosis (AS).
Methods and results: Eight patients with critical AS underwent temporary implantation of the PAV. Two patients received open surgical implantation of the device while six patients underwent percutaneous implantation. The mean age of these eight patients was 58.1 years, mean pre-procedural aortic valve area was <0.65 cm2 and pre-procedural gradient was 87.6±12.4 mmHg. Procedural success was achieved in seven out of eight patients, with a mean post-implantation gradient of 17.9±9.1 mmHg. There was one death related to inferior hypogastric artery dissection. All other patients subsequently received open surgical explantation of the PAV and aortic valve replacement with a mechanical valve.
Conclusions: The Direct Flow Medical PAV is technically feasible and safe to deploy in humans with severe AS and results in a significant transaortic gradient reduction.
Introduction
Surgical aortic valve replacement (AVR) remains the standard of care for patients with symptomatic aortic stenosis. However, open heart surgery caries a significant risk of mortality and morbidity. In patients undergoing isolated AVR, various registries have reported operative mortality rates ranging from 3.3% in New York State1 to 4.0% for the Society of Thoracic Surgery (STS)2. Among patients with multiple comorbidities such as endstage renal failure, severe pulmonary disease, severe heart failure, or prior CABG, these risks may be prohibitive for both the patient and surgeon. Over the last few years, percutaneous aortic valve replacement prostheses and techniques have been developed and provide hope for such high risk patients. The Edwards-Cribier and the CoreValve percutaneous valves have been well reported in the literature and represent the first generation of percutaneous aortic valves3-6. In this report, we describe the safety and feasibility using a second next generation novel technology, the Direct Flow Medical percutaneous aortic valve (PAV) from Direct Flow Medical, Inc., Santa Rosa, CA, USA.
Methods
Description of device
The Direct Flow Medical Percutaneous Aortic Valve (PAV) system is comprised of three key components, the bovine pericardial tissue heart valve, the sheathed delivery system with integrated recovery system and solidifying inflation medium (IM) that forms the support structure7.
Implant
The Direct Flow Medical PAV is designed as a durable surgical valve with anti-calcification treatment to the bovine pericardial leaflets. The tri-leaflet tissue valve is attached to an inflatable framework with a conformable polyester fabric cuff to provide a seal and minimise paravalvular leak. The implant is made functional initially by inflation with saline and contrast; once satisfied with the position and haemodynamic performance, the saline contrast mixture is exchanged for a solidifying IM that hardens to form the permanent support structure. The implant is designed in an hourglass shape, with independent inflatable ventricular and aortic rings which encircle and trap the native annulus to securely anchor the device. The PAV is available in multiple sizes (23 mm and 25 mm) (Figure 1).
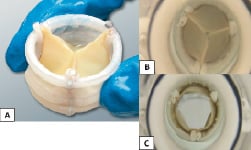
Figure 1. A: Direct Flow Medical Percutaneous Aortic Valve (PAV). B: Top view in haemodynamic tester during diastole. C: Top view in haemodynamic tester during systole.
The Direct Flow Medical PAV was initially tested in ex vivo pulse duplicator models. Valve durability passed over 200 million cycles in accelerated wear testing. In animal models, the PAV has been successfully deployed in more than 60 sheep with 18 sheep as chronic implant models with histological and pathological data to 365 days.
Solidifying Inflation Medium (IM)
The Direct Flow Medical PAV system utilises contrast and saline to initially expand the valve. When the final implant position is determined, the contrast and saline are exchanged with the proprietary IM. The IM is a biocompatible, two component liquid containing a water soluble epoxy and a radiopacifier. When mixed, the IM solidifies to a polymer in situ to maintain radial force and the position of the device within the native aortic valve. The IM in the liquid form is water soluble and has been tested to be non-embolic if released into the blood stream inadvertently.
Delivery system
The Direct Flow Medical delivery system is a 15 Fr catheter with an outer sheath at the distal end that houses the implant. The distal portion of the outer sheath is 22 Fr. The catheter contains three position/fill lumens (PFLs) which are attached to the implant. Two of these PFLs are used to inflate and deflate the cuff and all three are used to position the implant precisely in the native annulus. This enables positioning and repositioning of the implant. Integral to the delivery system is a recovery system which permits smooth retrieval through a 22 Fr introducer sheath. The entire delivery system is delivered over a 0.035” guidewire. (Figure 2)

Figure 2. Direct Flow Medical Delivery System. A: Sheathed. B: Unsheathed, prior to inflation. C and D: Inflated valve prior to detachment from delivery device.
Implantation technique
The first two patients underwent open surgical placement of the PAV in phase 1 as described below. Percutaneous delivery of the device was performed in subsequent patients. First, a 22 Fr introducer sheath was inserted in the femoral artery via a surgical cut-down. Prior to the procedure the implant was loaded into the Direct Flow Medical delivery system. Balloon valvuloplasty was performed using standard techniques to ensure adequate separation of the native calcified aortic valve leaflets. The valvuloplasty improved flow across the aortic valve and provided an increased orifice area for catheter delivery and PAV placement..
The constrained valve is then advanced to the native valve location over a super stiff 0.035” guidewire. Once the delivery system is in the left ventricle, the outer sheath is retracted exposing the implant. The ventricular ring is inflated independently from the aortic ring with the entire implant located in the left ventricular outflow tract. At this point, the valve is functional. The initial deployment of the inflatable cuff is accomplished by injecting contrast and saline (50/50) through the inflation PFLs to 10 atmospheres of pressure with a standard indeflator. The device is then retracted until the ventricular ring is seated and well sealed against the native annulus. Rapid pacing or cardiac support is not required during device placement. The aortic ring is then positioned above the annulus and inflated with a standard indeflator with contrast and saline. The position of the PAV relative to the left ventricular outflow tract, native valve and coronary ostia are depicted in Figure 3.
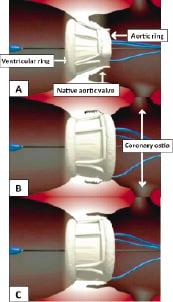
Figure 3. Graphic representation of the Direct Flow Medical PAV in relation to the coronary ostia at various stages of deployment. A: PAV with ventricular ring inflated. B: PAV with both aortic and ventricular ring inflated. C: Final position of PAV with both rings fully inflated.
If the device is not in the optimal location or if the initial size selection is not adequate as confirmed via fluoroscopy and echocardiography, the contrast and saline in both the ventricular and aortic rings can be withdrawn within seconds, thereby deflating the device. The device can be partially deflated and manipulated using the PFLs until optimal positioning is achieved. If final positioning is not achieved with optimal outcomes the device may be removed from the patient using the retrieval basket which is integral to the delivery system.
Once position and function are confirmed via fluoroscopy and echocardiography (Figure 4), the IM is introduced into the pressurised implant through the PFLs, displacing the contrast and saline out of the polyester cuff and back through the proximal end of the delivery catheter.
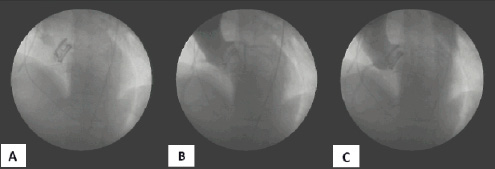
Figure 4. Flouroscopic images of the temporary deployment of the Direct Flow Medical PAV with 50% saline and 50% contrast mixture. A. Post valve expansion, pre-aortogram. B. Aortogram during coronary filling phase. C. Late aortogram demonstrating no paravalvular leak.
This process ensures there are no voids or air bubbles remaining in the implant. During the IM exchange the valve remains fully pressurised, in position, and competent. The implant is then detached from the delivery system and the delivery system is removed from the patient. Note that the PAV in this study was inflated with only saline/contrast media in the aortic and ventricular rings as surgical explantation was planned within 60 minutes.
A simultaneous pressure gradient is measured using the catheter guidewire lumen (left ventricular pressure) and a pigtail catheter (aortic pressure). Aortic root angiography and transesophageal echocardiography are then repeated to reassess valve competency and coronary patency. Angiographic aortic insufficiency severity was the consensus grade of two senior angiographers using standard criteria. Echocardiographic aortic insufficiency severity was the consensus grade of two senior echocardiographers.
Study design
The study was designed to evaluate the technical feasibility, safety and performance of the Direct Flow Medical PAV in patients with severe aortic valve stenosis using a controlled clinical evaluation to minimise patient risk and conclude with the retrograde percutaneous permanent placement of the PAV.
This report represents a series of human investigations: (1) open surgical implantation of the Direct Flow Medical PAV under direct visualisation and (2) temporary percutaneous implantation of the device. Phases 1 and 2 were conducted at The Hospital Privado Frances, Ascuncion, Paraguay. All clinical protocols in this study were approved by the Institutional Ethics Committee and prior to any procedure all patients provided written informed consent.
In phase 1, two patients underwent direct open surgical implantation of the PAV primarily for safety reasons. In these initial cases the patient was placed on cardiopulmonary bypass, the aorta opened and an intraoperative balloon aortic valvuloplasty was performed. The PAV was then positioned, deployed and detached from the catheter delivery system. After direct visual assessment of the PAV positioned in the native annulus, the aorta was surgically closed and the patient was subsequently taken off cardiopulmonary bypass for approximately 60 minutes. During this “temporary” deployment of the PAV, acute haemodynamic and echocardiographic measurements were recorded and the stability of the device in the aortic annulus was assessed. Figure 5 shows the final transesophageal and fluoroscopic appearance of the PAV.
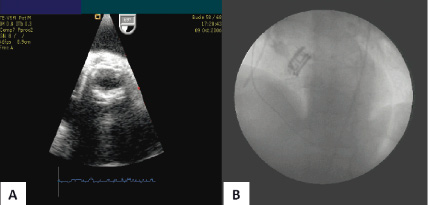
Figure 5. A. Post-procedure transesophageal echocardiography of the functional PAV. B. Post-procedure fluoroscopic appearance of the PAV.
Subsequently the patient was placed back on cardiopulmonary bypass and the PAV was surgically removed. A permanent Carbomedics mechanical valve (Carbomedics, Austin, Texas, USA) was then implanted using standard surgical AVR techniques.
Following this initial experience and the successful deployment of the first two devices via the open surgical technique, phase 2 implantations were performed using a standard transfemoral technique with the Direct Flow Medical delivery system in six patients. The objective of this phase was to confirm the Direct Flow Medical PAV and delivery system would perform as designed via a “percutaneous” delivery technique. A major goal of this study was to confirm the positioning and accurate deployment of the PAV via fluoroscopic imaging and if required the repositionability of the device in vivo. After 60 minutes of implantation, the patients underwent planned open surgical explantation of the PAV and surgical AVR with a Carbomedics mechanical aortic valve. Of note, two different generations of the device were tested in phase 2, with minor modifications in the latter generation. These modifications include adjustments in the diameter and height of the device. The Direct Flow Medical PAV utilised in this first human use experience in Paraguay had an overall height of 14 mm for both the 23 mm and 25 mm devices. It was observed during the initial phase of the study that the height of the implant was required to be greater than the native valve calcification. These observations led to dimensional changes to the current generation device with an overall height of 16 mm for the 23 mm; and 17 mm for the 25 mm device in order to accommodate a severely calcified annulus.
Based on the findings of the temporary implantations in Paraguay the implant underwent final design modification and complete design verification including preclinical, bench and durability testing. This initial feasibility study did not include a core angiographic or echocardiographic laboratory.
Study protocol
Subjects
Inclusion criteria required the following: (1) symptomatic severe aortic stenosis with an aortic valve area < 0.8 cm2 verified by transthoracic echocardiographic and Doppler imaging; (2) valvular and peripheral anatomy appropriate to the implant and delivery system; (3) patient age > 50 years; (4) aortic valve annulus diameter of > 21 mm and < 25 mm; and (5) suitable candidate for elective surgical aortic valve replacement.
Exclusion criteria include: (1) arterial access site unable to accommodate the 22 Fr introducer system due to size or tortuosity; (2) chronic renal sufficiency; (3) presence or suspicion of systemic infection; (4) contraindications to any study medications; (5) untreatable bleeding diathesis; (6) abnormal calcium metabolism (e.g., chronic renal failure, hyperparathyroidism); (7) hypercoagulable state; (8) congenital degenerative collagen disease (e.g., Marfan’s Syndrome); (9) prior valve or any cardiac surgery; (10) endocarditis; (11) acute cardiac decompensation; (12) severe mitral insufficiency; (13) severe aortic insufficiency; and (14) stroke in the past six months.
Outcome measures
The primary outcome of phase 1 and 2 is procedural success which is defined as accurate placement of the valve in the subcoronary position with associated improvement of the trans-aortic gradient and absence of severe aortic insufficiency. Safety is determined by the absence of major adverse cardiac and cerebrovascular events (MACCE) or other valve-related adverse events. MACCE in this protocol is defined as death, myocardial infarction, cerebrovascular accident or dislodgement or embolisation of the PAV.
Results
Nine patients were eligible and consented for Phases 1 and 2. The procedures were performed in October 2006 and March 2007.
Of the nine patients from phases 1 and 2, eight had successful implantation of the device. In one patient, one of the PFLs became dislodged prematurely. The patient did not receive the device and instead underwent successful surgical aortic valve replacement. Therefore, a total of eight patients received the PAV and will constitute the basis of the results (Table 1).
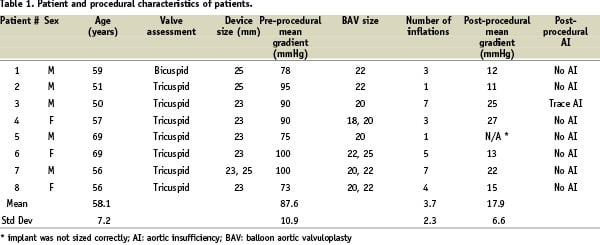
In one patient, an inappropriately sized device was used resulting in procedural failure. Therefore, procedural success was achieved in seven of eight patients. The mean pre-procedural aortic valve area was 0.65 cm2 and pre-procedural gradient was 87.6±12.4 mmHg. The mean post-procedural aortic valve area in the seven successful patients was 1.65 cm2 and post-procedural gradient was 17.9±9.1 mmHg. Immediate post-procedural evaluation showed that in seven of the eight patients, there was no aortic insufficiency noted on transesophageal echocardiography and fluoroscopy. In one patient, there was trace aortic insufficiency. In 2 patients, the initial implant size was felt to be too small and the implants were retrieved successfully. Larger implants were then successfully delivered.
There were two instances of vessel dissection. The first was caused by advancement of the guidewire, resulting in perforation of the external iliac artery. This was immediately recognised and treated by advancing the sheath further into the vessel. The PAV was successfully deployed and the artery was subsequently repaired during the surgical component of the study. The second involved dissection of the inferior hypogastric artery, and was thought to be related to advancement of the introducer sheath. This was not initially recognised. The diagnosis was made subsequently and the patient underwent emergent surgical repair. Despite aggressive resuscitative efforts, the patient expired.
Discussion
This proof-of-concept report on the implantation of the Direct Flow Medical PAV demonstrates the device can be safely implanted percutaneously with excellent acute haemodynamic results. This series of studies; two implants under direct visualisation and six percutaneous temporary implantations verified the functionality of the catheter delivery system, the integral retrieval system and the acute implant performance of the Direct Flow Medical PAV.
The methodology utilised in these evaluations demonstrates the acute haemodynamic profile of the PAV and its ease of deliverability in humans. Importantly, the study provided proof of concept that an inflatable cuff could provide sufficient support to hold the PAV in place. Second, the PAV, which conforms to the aortic annulus, provides a significant reduction in the trans-aortic pressure gradient with minimal or no aortic regurgitation. These methods also permitted rapid product development from design concept to human clinical evaluation, with important design feedback. For example, a patient in the phase 2 evaluation developed a post-implant gradient of 50 mmHg. This gradient was determined to be a result of the device height being less than the native leaflet length. This design input resulted in a valve height enhancement (described earlier) which has improved the performance of the device. The increased device height was only 2 and 3 mm respectively for the 23 and 25 mm devices respectively and was designed to cover only the native valve leaflet length and calcification. Therefore, the increased height was not expected to extend to the level of the coronary ostia. Nonetheless, the Direct Flow Medical PAV allows real-time evaluation of device positioning. Aortography and coronary angiography can be performed with the device deployed to ensure patency of the coronary ostia. The device can be adjusted if necessary or removed if an ideal position cannot be achieved.
In this series there was one patient death in the Phase 2 study. This death was related to the insertion of the introducer sheath which caused an unrecognised arterial dissection. The need for large arterial sheaths to introduce PAV devices has been an important consideration in the selection of eligible patients. Patients with small ilio-femoral vessels, tortuous anatomy or severe calcifications are generally excluded from trials involving PAV4,5. In a series of patients with the Edwards prosthesis, Webb et al reported that the first two patients had similar iliac arterial complications with one subsequent death3. In this feasibility study, peripheral arterial anatomy was assessed pre-procedure but not with the specification that is currently employed. The feasibility study demonstrated severely calcified, tortuous vessels and arteries <7 mm would constitute contraindications to the use of this early generation catheter. Future iterations of the device will incorporate advancements that will allow its use in more challenging patients. Nonetheless, our experience and that reported in the literature highlight the critical importance of access site evaluation and exclusion of unsuitable patients.
The advantages of this device include the following. First, the innovation using the IM allows the supporting structure of the PAV to be formed in situ. This feature permits the device to be re-positionable and retrievable after initial deployment. Only after the IM is exchanged and the device is released does the implantation become permanent. This is an important advancement in the field as a major limitation of previous devices is that they can neither be re-positioned nor retrieved once deployed. It also allows for the evaluation of the optimal deployment and re-deployment of the PAV if necessary. This is a key advantage given the proximity of the coronary arteries to the native valve and the importance of creating a good seal to the native aortic valve and left ventricular outflow tract. Second, delivery of the device is standard over a 0.035” stiff guidewire and requires no special controller or guide catheter to negotiate the aortic arch. This is due to the non-metallic construction of the PAV and the highly trackable delivery catheter. Third, the polyester cuff containing the inflatable rings is very conformable, and when expanded with IM, conforms to the native anatomy, thus reducing paravalvular aortic insufficiency which is sometimes associated with the Edwards-Cribier and CoreValve PAVs4,5. Fourth, the device does not require cardiac support or rapid pacing during deployment or positioning.
The potential disadvantages of the device include the following. First, the use of PFLs, while giving the operator great control over the positioning of the device, also requires careful manipulation. Second, compared to the Edwards-Cribier and CoreValve PAVs, which utilise primarily an interference fit of the metal framework against the calcified annulus / leaflets to maintain their position; the Direct Flow Medical PAV relies on primarily the ‘sandwiching’ effect of the ventricular and aortic rings to encapsulate or ‘grip’ the native valve to maintain position and seal. Because of that, sizing of the device becomes important, as undersizing of the device may result in slippage.
The early experience with this device indicates it can be safely and effectively deployed, with excellent haemodynamic outcomes. The ability to be able to non-invasively retrieve the device is an important advancement to the field of percutaneous aortic valve replacement. However, the two vessel perforations that occurred, one of which resulted in a death, demonstrate the importance of pre-procedural evaluation of the extent of peripheral arterial disease to ensure appropriate anatomy and patient selection. Physicians performing percutaneous PAV should maintain a heightened sense of awareness of the risk for aorto-ilio-femoral injury. Future iterations of this device are expected to include smaller diameter delivery catheters.
In summary, the Direct Flow Medical PAV is a second generation repositionable and retrievable PAV device that has been shown to be easily deployed in man. It is a novel addition to the field of percutaneous heart valve therapy. Ongoing European trials will provide further information on the feasibility, safety and long term outcomes of this device.
